|
Biodiversity Data Journal :
Methods
|
|
Corresponding author: Peter M Hollingsworth (phollingsworth@rbge.org.uk)
Academic editor: Gergin Blagoev
Received: 18 Feb 2023 | Accepted: 04 Aug 2023 | Published: 27 Oct 2023
© 2023 Giada Ferrari, Lore Esselens, Michelle Hart, Steven Janssens, Catherine Kidner, Maurizio Mascarello, Joshua Peñalba, Flávia Pezzini, Thomas von Rintelen, Gontran Sonet, Carl Vangestel, Massimiliano Virgilio, Peter Hollingsworth
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation:
Ferrari G, Esselens L, Hart ML, Janssens S, Kidner C, Mascarello M, Peñalba JV, Pezzini F, von Rintelen T, Sonet G, Vangestel C, Virgilio M, Hollingsworth PM (2023) Developing the Protocol Infrastructure for DNA Sequencing Natural History Collections. Biodiversity Data Journal 11: e102317. https://doi.org/10.3897/BDJ.11.e102317
|
|
Abstract
Intentionally preserved biological material in natural history collections represents a vast repository of biodiversity. Advances in laboratory and sequencing technologies have made these specimens increasingly accessible for genomic analyses, offering a window into the genetic past of species and often permitting access to information that can no longer be sampled in the wild. Due to their age, preparation and storage conditions, DNA retrieved from museum and herbarium specimens is often poor in yield, heavily fragmented and biochemically modified. This not only poses methodological challenges in recovering nucleotide sequences, but also makes such investigations susceptible to environmental and laboratory contamination. In this paper, we review the practical challenges associated with making the recovery of DNA sequence data from museum collections more routine. We first review key operational principles and issues to address, to guide the decision-making process and dialogue between researchers and curators about when and how to sample museum specimens for genomic analyses. We then outline the range of steps that can be taken to reduce the likelihood of contamination including laboratory set-ups, workflows and working practices. We finish by presenting a series of case studies, each focusing on protocol practicalities for the application of different mainstream methodologies to museum specimens including: (i) shotgun sequencing of insect mitogenomes, (ii) whole genome sequencing of insects, (iii) genome skimming to recover plant plastid genomes from herbarium specimens, (iv) target capture of multi-locus nuclear sequences from herbarium specimens, (v) RAD-sequencing of bird specimens and (vi) shotgun sequencing of ancient bovid bone samples.
Keywords
Museomics, hDNA, biodiversity genomics, natural history collection sequencing
Introduction
Natural history collections as a resource for genomic science
There are more than one billion specimens representing ca. two million species stored in natural history collections worldwide (
Until recently, the recovery of DNA sequences from museum specimens was challenging and prone to very high rates of failure or requiring laborious protocols for successful recovery of minimal quantities of nucleotide sequence data (
Storage and preservation of museum specimens
The global collection of preserved natural history specimens contains a diverse set of samples encompassing a multitude of different tissue types and preservation methods (
Properties of DNA in natural history collections and implications for sequencing museum specimens
The DNA within natural history museum specimens has distinct properties from the DNA in freshly-collected material which has practical implications for recovering sequence data. From a biochemical perspective, DNA isolated from natural history collection specimens shares many similarities with ancient DNA (aDNA). Characteristically, aDNA is highly fragmented and biochemically damaged, often present in small quantities and subject to contamination from the environment and human handling. In the absence of the enzymatic repair mechanisms of living cells, DNA is subject to hydrolysis, oxidation and cross-linking (
These degradation processes for aDNA also occur to greater or lesser degrees in museum specimens. Various studies of DNA degradation in natural history collections have shown that DNA fragmentation can occur rapidly after death (
Following the fragmentation of DNA in museum specimens, there is a consequential and associated loss of DNA as DNA fragments diffuse away from specimens.
There are several consequences of this fragmentation and loss of DNA. The first is that experimental effort may be expended which ultimately leads to a failure to recover DNA sequence data due to low endogenous DNA content. The second is that DNA sequence data may be recovered, but be misleading due to contamination. The potential for contamination in sequence data from museum specimens is substantial. The low concentrations of fragmented endogenous DNA in museum specimens represent an initial low signal-to-noise ratio and a high potential for contamination from a wide variety of sources, including:
-
Biological material on the specimen (surface contaminants and biological materials associated with specimen preparation);
-
High concentrations of DNA from fresh samples and their amplification products processed in the same facility which represent an important source of contamination when handling degraded DNA. Such contaminant DNA may be present in higher concentrations than the DNA in historic samples and this is exacerbated by subsequent PCR being biased towards higher-quality DNA;
-
General contamination in the processing lab, including sources of contaminating DNA from specimen handling, laboratory reagents and aerosols in the wider environment.
At best, contamination reduces sequencing efficiency for endogenous DNA and requires greater sequencing efforts at higher costs. What is more problematic is the generation of erroneous data where misleading biological inferences are made from undetected contamination (
A more generic source of error, but one to which museum-derived sequences are particularly susceptible, is problems stemming from low coverage of sequence reads due to low DNA concentration. This can result in misleading inference; for example, failure to recover both alleles in diploid heterozygotes leading to an overestimation of homozygosity at some loci and in some specimens (
Above and beyond the challenges of recovering reliable sequence data from low concentrations of fragmented endogenous DNA is the possibility of post-mortem modifications leading to artefactual substitutions in the recovered DNA sequences. Post-mortem hydrolytic deamination causes base modifications, primarily affecting cytosine (
Outstanding challenges to the routine sequencing of museum specimens
There has been a recent shift in the field of museomics from small-scale studies, often with high rates of failure, to increased success rates and a growth of increasingly-ambitious studies aiming to liberate sequence data at a large scale from museum collections (
-
Deciding when it is appropriate to sample museum specimens: Development of guiding principles to facilitate sampling decisions that support specimen utilisation, but avoid unnecessary and unproductive destructive sampling;
-
Minimising the risks of contamination and production of erroneous sequence data: Guidance and utilisation of appropriate laboratory infrastructure and data verification steps;
-
Maximising the recovery of endogenous DNA sequences: Optimisation of protocols to improve the efficiency and efficacy of different widely used techniques.
These three topics are addressed in subsequent sections of this paper.
Deciding when it is appropriate to sample museum specimens for DNA sequencing
Destructive sampling poses a dilemma between damaging a specimen for research utilising existing protocols and preserving the specimen for future and improved methodologies (
To minimise damage to specimens, a number of minimally or non-destructive sampling protocols for collection material have been proposed. DNA extraction protocols for ancient and historic DNA have been optimised to obtain good DNA yields from small amounts of material, for example, as low as two milligrams of dry plant tissue (
Maximising the use of museum specimens, including for genomic analyses, while minimising unnecessary destruction of precious samples thus reflects a balancing act. This is made particularly difficult, as often the greatest scientific returns will come from the specimens that are most valuable. For instance, type specimens will almost always have significant constraints on their use, which may act as a barrier to inclusion in genomic studies. On the other hand, effective minimally destructive sequencing of type specimens provides a direct connection between genomic data and the application of a species name and, hence, represents a significant scientific benefit, particularly for taxonomic and systematic studies. There is a general point, that while sampling a museum specimen for genomic analysis usually results in something being taken away from the specimen to obtain DNA, it can also result in something extremely useful being added, in terms of critically important genomic data which may add considerable value to the specimen (i.e. the concept of the extended specimen;
Guidance on best practice standards and processes for the access and transfer of samples for genomic analysis is given by the
- Assess the scientific merit of the planned genomic project; ensure there is a clear likely benefit prior to commencing destructive sampling and that the resulting data will be informative and of sufficient resolution to tackle the question at hand;
- Always adopt a minimally destructive approach for genomic studies of museum specimens unless there is a clear surplus of available tissue, such as extensive duplicate specimens or ‘sacrificial’ specimens available for experimentation;
- Utilise alternative options to destructively sampling important specimens if available (e.g. make use of any reliable previously-sampled tissues or previous DNA extracts, adopt a non-destructive sampling approach, if applicable);
- If multiple tissue types are available, consider the likely success rates for the different tissues and weigh this against their respective morphological impacts on the specimen in choosing which tissue to sample;
- Seek to maximise the reusability of the data from destructive sampling: consider methodologies which give maximum amounts of data which will be of use for multiple downstream applications;
- Seek to maximise the reusability of DNA from destructive sampling: adopt methods of handling and storing DNA extracts to maximise their preservation and reuse potential to minimise further need for specimen sampling;
- Process samples following appropriate laboratory controls and with clear data-verification steps to ensure that the resulting data have maximum reliability and value;
- Evaluate the feasibility of success prior to destructive sampling of valuable specimens in terms of protocol efficacy, researcher capability and laboratory suitability and only proceed where the likely chances of success and resulting scientific benefits outweigh the costs of any destructive sampling;
- Report successes and failures to guide future optimisation of protocols and decision-making regarding destructive sampling;
- Ensure appropriate accessibility of the resulting sequence data and linkages and connections between the data and the specimens they were derived from, to ensure that specimen sampling for genomic analyses results in added value to the specimen itself.
Minimising the risks of contamination and generation of erroneous sequence data
Historic DNA versus ancient DNA versus modern optimally-preserved DNA
The opportunities arising from the sequencing of museum specimens have attracted researchers from different backgrounds and fields. On the one hand, sequencing the degraded DNA in museum specimens has long been a focus for aDNA researchers. Ancient DNA techniques involve working with low concentrations of highly-degraded DNA in specialist laboratories with strict guidelines and meticulous anti-contamination precautions (
The DNA in the majority of natural history museum specimens sits at the interface of aDNA and non-degraded DNA samples and is classed as historic DNA (hDNA), more formally defined as DNA from specimens archived in museum collections that were not originally intended as genetic resources (
Current operational practices for processing hDNA
The maximally effective recovery of hDNA is dependent on determining the appropriate levels of stringency of laboratory practices, which minimise risks of contamination, while at the same time, being sufficiently scalable to allow maximum utilisation of the vast resources of specimens available in museum collections. Thus, while utilising dedicated aDNA facilities (
During the preparation of this paper, discussions amongst the authors and an informal survey of colleagues working in a range of organisations involved in sequencing museum specimens, revealed a wide range of operational practices. These ranged from processing samples in the same laboratories as fresh tissue, through to dedicated hDNA (or low-copy) laboratories, through to only ever using fully-equipped aDNA facilities for processing museum specimens. A multitude of factors were articulated as underlying the decision-making of which facilities to use for processing hDNA samples, including:
-
Resource constraints (money and/or space) precluding establishment of a dedicated facility;
-
Desire to use existing facilities in standard labs to enable processing of large numbers of samples;
-
Controls in place for data authentication and/or stringent cleanliness conditions in standard labs considered adequate to negate the need for a dedicated facility for hDNA samples;
-
Individual preferences of researchers determining where samples are processed without clear institutional policies;
-
When both aDNA and hDNA samples have to be processed in the same institution, the aDNA laboratory being used exclusively for aDNA samples, with museum specimens processed elsewhere due to concerns that the higher concentrations of DNA from museum specimens may lead to contamination problems in the aDNA lab.
Laboratory set-ups and workflows for hDNA sequencing
A general observation noted by several researchers familiar with working with non-degraded DNA samples was a lack of clarity over what an optimal laboratory set-up would look like for hDNA analysis. To facilitate evaluation of options for contamination control and the practicalities of laboratory set-ups, we outline below the headline infrastructure and working practices of an aDNA facility and a basic hDNA facility. We also list contamination-limiting recommendations for processing degraded material in existing, more general, laboratories.
Ancient DNA facility
The most critical component of setting up an aDNA laboratory (
Historic or low-copy DNA facility
Natural history collection material can be handled in aDNA laboratories. However, aDNA laboratories and their upkeep can be prohibitive in cost; therefore, institutions working exclusively on natural history collections may choose a less stringent set-up for a dedicated hDNA pre-PCR facility. This would usually be located in existing rooms rather than a purpose-built laboratory. Not requiring a positive pressure air system and a laboratory antechamber allows for more flexibility in the choice of location (e.g. repurposing of laboratory spaces) and significantly reduces the costs. It should be noted that if an hDNA facility is being established by repurposing an existing laboratory, thorough cleaning with sodium hypochlorite of all surfaces is essential and new, dedicated equipment should be bought. Similarly to aDNA facilities, all work should take place inside UV-fitted PCR cabinets, and destructive sampling and sample powdering should take place in a separate room or at least in a separate cabinet. Additional UV lamps for surface decontamination may be fitted; however, repeated UV exposure is damaging to laboratory equipment, increasing upkeep costs. Cleaning routines and good practices as described for aDNA facilities should be implemented or adapted as best as possible, most importantly the separation of pre- and post-PCR working areas. In Suppl. material
Contamination-limiting measures for working in existing facilities
Due to space or financial constraints or because of the need for higher throughput, it remains the case that many institutions may decide against an aDNA or hDNA facility and process natural history collection material in existing laboratories alongside fresh biological material. Fresh material and especially its amplification products, represents a common source of contamination for historic material. Thus, the separation of pre- and post-PCR areas remains essential, although the routes to achieving this can be varied. At the very least, thermocyclers should always be located in the post-PCR area and movement of samples, reagents, consumables and equipment between pre- and post-PCR should be avoided or limited as best as possible. Additionally, pre-PCR work on collection material should be carried out in dedicated laminar flow hoods (to be cleaned and UV-sterilised regularly) with dedicated tools and reagents.
Data verification and contamination controls
In addition to the physical layout of laboratories, data verification and contamination controls include:
Verification checks against reference libraries: The likelihood of being misled by contamination in the sequencing of museum specimens is a function of the stringency of the controls and the complexity of the detection task. Data-authentication steps can be relatively straightforward, where there is an a priori expectation of the sequence to be recovered and an existing reference resource to check it against. Thus, studies like large-scale DNA barcoding projects, are intrinsically well-suited to contamination checks, with small regions of DNA being recovered from individual museum specimens which are usually identified to species level and whose identity can be checked by sequence cluster placement in existing DNA barcode reference libraries such as BOLD (
Ordering samples to avoid closely-related taxa being in adjacent wells: The most difficult contamination to spot is from closely-related samples, as even detailed analysis and comparisons with reference samples may not flag contaminants. Where there is a mixture of closely- and more distantly-related taxa being processed, a simple option is to order samples to maximise the likelihood of adjacent well contamination being detectable, by minimising the presence of closely-related specimens in adjacent wells.
Negative controls: The inclusion of non-template negative controls at the DNA isolation and library preparation/PCR step is essential for ensuring that the pre-PCR facilities and reagents are sufficiently clean. Negative controls should also be taken through all post-amplification steps, sequenced and included in the data analysis.
Sequencing strategies: Jumping PCR (
DNA repair: To mitigate against misleading inference due to post-mortem DNA modifications, enzymatic treatment of DNA extracts can be undertaken, for instance with the USER reagent (New England Biolabs), a mix of uracil–DNA–glycosylase (UDG) and endonuclease VIII (
Data authentication and validation: In addition to checks against reference libraries, various bioinformatic pipelines and workflows support the verification and authentication of degraded DNA sequences. These include estimating contamination by screening for unexpected results such as 'heterozygosity' of haploid genomes (mitogenomes, plastomes) (
Maximising the recovery of endogenous DNA sequences
Over the last decade, there has been a constantly expanding set of literature outlining new developments which contribute to making the recovery of sequence data from museum specimens more cost-effective and routine (
The generation of guidelines and decision-making frameworks to support more routine recovery of sequence data from collections is of considerable value as the field of museum collection sequencing expands (
Table
Selected papers outlining recent progress, breakthroughs and protocol developments that support the more routine recovery of genomic data from museum specimens
|
Study taxon |
Tissue type |
Specimen ages (yrs) |
Approach(es) |
Key finding |
Reference |
|
Plants |
Dried herbarium specimens |
2-182 |
Multi-locus nuclear sequence capture |
Large-scale study of 7608 specimens using angiosperm 353 target capture baits; DNA yield poor predictor of sequencing success, plant family strongest predictor of success, successful recovery of old specimens from tropical climates |
|
|
Plants |
Dried herbarium specimens |
NA |
NA |
Protocols and best practices for working with ancient and historical plant DNA. Includes laboratory set-up, DNA isolation, sequencing library preparation and bioinformatic analyses |
|
|
Plants |
Dried herbarium specimens |
Up to 280 |
Shotgun sequencing |
DNA in herbarium specimens degrades faster than in ancient bone. Both fragmentation and deamination accumulate over time |
|
|
Plants |
Dried herbarium specimens |
most < 20 yrs, 165 samples > 50 yrs; oldest 153 yrs |
Shotgun sequencing |
Large-scale genome skimming study of 2051 herbarium specimens recovering plastome and rDNA sequences including standard plant barcodes |
|
|
Fungi |
Rust fungi on dried herbarium specimens |
Up to 187 |
Amplicon-based rDNA sequencing |
Protocol development and application to track dynamics of plant pathogens through time sampled from herbarium specimens |
|
|
Fungi |
Fungarium specimens |
Less than 20 |
Whole genome sequencing |
Generation of draft genome assemblies possible and of value for enhancing resolution of fungal phylogeny |
|
|
Birds |
Avian skins |
up to ca. 150 |
Whole genome sequencing |
Step-by-step guide to workflow and protocols, including steps taken to minimise risks of contamination |
|
|
Mammals (mephitids, rodents, marsupials) |
Dried museum skins |
50-120 |
Shotgun sequencing |
Comparison of DNA yields and bacterial contamination levels in commonly-sampled museum mammalian tissues (bone, claw, skin and soft tissue) and implications for sampling strategies |
|
|
Mammals (grey wolf) |
Dried museum skins |
90 - 146 |
Shotgun sequencing |
“Single-tube” DNA library preparation methods including adaptations for degraded DNA increase library complexity, yield more reads that map uniquely to the reference genome and reduce processing time compared to other Illumina library preparation methods |
|
|
Mammals (bison and horse) |
Bone |
Up to 40,680 |
Shotgun sequencing |
A more accessible single-stranded genomic library preparation method optimised for aDNA |
|
|
Mammals (dogs and mammoths) |
Bone |
Up to 37,080 |
Shotgun sequencing |
Competitive mapping of raw sequencing data to a concatenated reference composed of the target species' genome and the genome of possible contaminants contributes to filtering out contamination from ancient faunal DNA datasets with limited losses of true ancient data |
|
|
Mammals (Cricetidae, rodents, deer mouse) |
Frozen liver tissue |
17 to 41 |
Whole genome sequencing |
Linked-read or “synthetic long-read” sequencing technologies provide a cost-effective alternative solution to assemble higher quality de novo genomes from degraded tissue samples |
|
|
Insects (Phylinae; plant bugs) |
Abdomen |
1 to 54 |
DNA bait capture |
Inexpensive data generation to produce sufficient amount of data to assemble the nuclear ribosomal rRNA genes and mitochondrial genomes |
|
|
Insects (Apidae, bumble bees) |
Leg |
18 to 131 |
Shotgun sequencing |
DNA in entomological specimens in NHC highly degraded, process age dependent with a roughly linear reduction in fragment length over time after strong initial fragmentation |
|
|
Insects (Culicidae, mosquitoes) |
Whole specimens |
33 to 84 |
Shotgun sequencing |
Minimally damaging extraction method for building libraries for Illumina shotgun sequencing |
|
| Insects (Lepidoptera) | Leg | Average 50 years | Sequel CO1 barcoding | Demonstration of utility of PacBio Sequel platform for recovery of full length CO1 barcodes |
|
A general challenge for the effective recovery of endogenous DNA from museum specimens is the frequent low complexity of libraries caused by PCR and cleaning steps modifying the relative abundances of the original DNA fragments during library preparation (
Case study overview
To provide further details on protocol development for particular genomic approaches and their success in application to different taxonomic groups, we present a series of case studies undertaken at different institutions as part of the EU SYNTHESYS+ project. Each focuses on protocol practicalities for the application of different mainstream methodologies to museum specimens including: (i) shotgun sequencing of insect mitogenomes (Museum für Naturkunde Berlin), (ii) whole genome sequencing of insects (Royal Museum for Central Africa), (iiii) genome skimming to recover plant plastid genomes from herbarium specimens (Meise Botanic Garden), (iv) target capture of multi-locus nuclear sequences from plants (Royal Botanic Garden Edinburgh), (v) RAD-sequencing of bird specimens (Royal Belgium Institute for Natural Sciences) and (vi) shotgun sequencing of ancient bovid bone samples (Royal Belgium Institute for Natural Sciences).
Case study 1: Assessing the potential of rapid shotgun sequencing for the recovery of mtDNA genomes from pinned insect specimens
Introduction
The world’s entomological collections hold more than half a billion pinned (dried) insect specimens (
This potential has received increasing attention in recent years (
Materials and Methods
Specimens (n = 70 in total) were selected from a genus (or two closely-related genera) from each of three major holometabolan insect groups (Diptera, Hymenoptera, Lepidoptera) with a collecting date ranging from 1891 to 2015 (132-8 years in collection storage). The taxa (Diptera: Sarcophaga; Lepidoptera: Eudonia, Scoparia; Hymenoptera: Xylocopa) are actively studied by the respective curators at Museum für Naturkunde and the availability of at least one reference mitogenome was an additional core criterion. One leg each was used for DNA isolation using the Qiagen Investigator Kit. Multiplexed libraries for paired-end Illumina sequencing were prepared for each sample with the “NEXTflex Rapid DNA Seq Kit 2.0” and “NEXTflex HT Barcodes” (Bioscientific/PerkinElmer). DNA input varied between 1 and 300 ng (10 ng or less for > 75% of samples). As the DNA of all samples was expected to be strongly degraded, the protocol was adapted accordingly (no shearing and bead-size selection, adjustment of bead buffer - sample ratio for clean-up after adapter ligation). Library quality, size and quantity were determined with a TapeStation (D1000 Kit Kit/ Agilent) and Qubit 2.0 Fluorometer (dsDNA HS Assay Kit/Thermo Fisher Scientific). Libraries were pooled, based on these parameters and low-coverage test sequenced on an Illumina Miseq (PE150). Based on the results from the test sequencing, 12 libraries were subjected to a booster PCR to increase quantity. Libraries were re-pooled, based on library parameters (concentration) and test sequencing results (number of reads) and the resulting pool was sequenced on an Illumina Nextseq (PE150). The cleaned and de-multiplexed reads were mapped against mtDNA genomes of the respective target taxon using the MITObim pipeline (
Results and Discussion
The amount of recovered DNA ranged from 3 ng to 1.2 µg (Fig.

Scatter plots of DNA and sequence recovery from pinned insect specimens by age and taxon (blue - Eudonia/Scoparia, grey - Sarcophaga, orange - Xylocopa). Specimen age (in years) is on the x axis in all panels. A Total DNA yield (ng). B Number of sequencing reads. C Completeness of the mitogenomes (%). D Coverage (n).
Success rate in relation to sample age
DNA degradation is affected by several variables, such as initial preservation of samples and storage conditions, which in themselves are highly diverse in many aspects (temperature, humidity, pest control chemicals). As the effect of these factors accumulates over time, an obvious assumption is that DNA degradation will be worse in older specimens and sequence recovery consequently more difficult, as has been shown in a recent systematic study using NGS methods (
Overall, no sample failed entirely as measured by obtaining reads from mtDNA. One sample did not yield any reads at all after the booster PCR, but the same sample worked (albeit with only ca. 81,000 reads) for the library prepared without additional PCR amplification.
In contrast to age, variation in success rate does not seem to differ amongst taxa, if variation amongst collecting dates is taken into account. Almost all samples of Xylocopa, for example, were collected before 1940 and about 75% in 1912 or before, and the apparently lower success rate for Xylocopa could be explained by age alone. Similarly, the species and consequently specimens used in this study differed in body size, which was not controlled for here and, as the total amount of DNA recovered is expected to be dependent on tissue input, that value cannot be directly compared meaningfully between specimens of different taxonomic groups.
Effectiveness in terms of costs and time of shotgun sequencing
Shotgun sequencing is a straightforward and technically undemanding approach by NGS standards. In recent years, it has also become ever more cost effective as measured by cost per bp. For example, in this study, each sample yielded on average 1.6 mio. reads and sequencing costs per sample were ca. €32 (price as of December 2022). These costs could be further reduced using different sequencing platforms (e.g. Illumina Novaseq).
Interestingly, to date, a limited number of studies have used shotgun sequencing for museomics in insects and only one has targeted mtDNA in particular for taxonomy in a specimen-based approach. All types of Australian prionine longhorn beetles were shotgun sequenced by
A frequently-used alternative to shotgun sequencing for recovering genomic data from museum specimens is target capture (e.g.
For both low-coverage shotgun sequencing and target capture, there is a trade-off between costs, time and sequencing success (measured by the completeness of the target sequence(s)), which is likely to tilt towards bait capture when the aim is to sequence a large number of closely-related samples. However, if the aim is to target a larger range of taxa at the genus level and beyond for taxonomic purposes, such as DNA barcoding, then shotgun sequencing has the edge in our opinion due to its relative ease and the universality of approach. As sequencing costs are still on a downward trajectory, the cost balance is likely to be tilted further in its favour in the future.
Main findings and recommendations:
-
A shotgun approach is particularly appropriate for obtaining (mtDNA) data for a wide range of different taxa with relatively little effort in the lab, which makes it highly useful for taxonomy and providing reference sequences from type material. If the aim is to generate complete datasets from many individuals of closely-related species, bait capture might be a viable alternative;
-
Older samples will often require more sequencing effort to obtain the same amount of data as more recent specimens. If the main aim is the generation of DNA barcodes for taxonomic purposes, this should not be overly relevant in practice;
-
When using a shotgun approach, using a leg is sufficient to obtain an adequate amount of data for taxonomic purposes at least from medium-sized to large (> 10 mm) specimens, which should make it easier for curators to give permission for destructive sampling;
-
Crucially, when adding to collections, sample preservation should be optimised in the field in order to avoid heavy DNA degradation before specimens become museum specimens.
Data availability: The raw sequencing data from this case study has been deposited at the European Nucleotide Archive (ENA) under project PRJEB59182 with accession IDs ERS14475133 - ERS14475206.
Case study 2: Genomic vouchering in insect museum collections: the quest for a pragmatic approach to routine, large-scale genotyping
Introduction
The costs directly related to genomic library preparation and sequencing represent one of the main limiting factors hampering the whole genome sequencing (WGS) of large numbers of museum specimens. Until recently, the partial sequencing of genomes, via approaches, such as reduced representation libraries (
Materials and Methods
Comparative performances of commercially-available DNA extraction kits
The performance of commercial DNA extraction kits (see below) was compared in a pilot study targeting the Royal Museum of Central Africa (RMCA) collections of “true” fruit flies (Tephritidae, Diptera) and African hoverflies (Syrphidae, Diptera). We selected three to six specimens from seven sample groups dating from 2008 to 2016. These included three Tephritidae (Zeugodacus cucurbitae Coquillett, Bactrocera dorsalis Hendel and Dacus bivittatus Bigot) and two Syrphidae (Eumerus sp. and Ischiodon aegyptius Wiedemann) species; all specimens were stored in 100% ethanol at -20°C, except Ischiodon aegyptius which was pinned and preserved at room temperature. Digestions in lysis buffers were implemented on whole bodies for all specimens. For comparative purposes, we also processed forelegs of a separate set of specimens. The lysates obtained from each specimen were divided into four aliquots and the DNA purified using spin columns from the DNA extraction kits listed in Table
An overview of the DNA extraction kits tested on fruit flies and hoverflies.
|
QIAGEN kit (50 samples) |
Spin column |
Range of DNA fragment sizes (according to manufacturer’s instructions) |
Expected DNA yield (according to manufacturer’s instructions) |
|
DNeasy Blood and Tissue Kit |
DNeasy spin column |
100 bp-50 kb |
6-30 µg |
|
QIAamp Micro Kit |
QIAamp MinElute column |
< 30 kb |
< 3 µg |
|
QIAamp Mini Kit |
QIAamp Mini spin column |
< 50 kb |
4-30 µg |
|
DNeasy Blood and Tissue Kit |
MinElute column (MinElute PCR Purification Kit) |
70 bp-4 kb |
< 5 µg |
Relationships between voucher DNA quality and WGS performance
To assess the relationship between WGS performance and (a) voucher age and preservation and (b) DNA quality and quantity, we targeted a total of 732 insect vouchers archived in the collections of RMCA collected from 1997 to 2022 (Fig.

Based on DNA concentrations (above or below 7.0 ng/µl) and DNA fragmentation (> 350 bp or highly fragmented defined as < 350 bp), samples were submitted to Berry Genomics (n = 563) for standard library preparation or to Novogene (n = 81) for low input DNA library preparation, respectively. All samples were sequenced at 10x coverage on an Illumina NovaSeq platform (150 PE reads, 6 Gb raw data output/sample). Quality parameters of extracted DNA and WGS data of specimens originating from five insect genera and more than 70 different species were collected (see Table
Results and Discussion
Overall, our DNA extractions provided detectable DNA in 720 specimens out of 732 (98.4%) while WGS data could be obtained for 644 museum vouchers (88.0%). The different DNA extraction methods gave broadly similar yields, albeit with a somewhat lower recovery of DNA from whole-body extractions using the MinElute kit. Overall, there was a heterogeneous recovery of DNA yields across specimens (Fig.
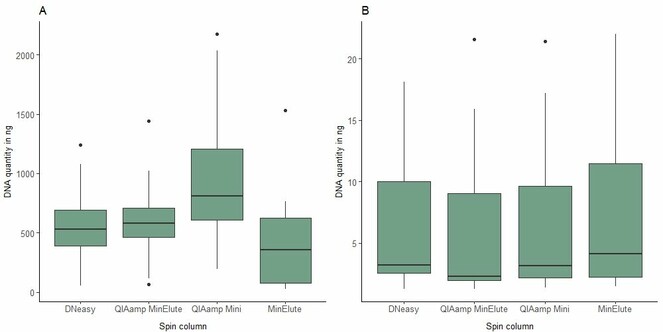
Boxplots of DNA yields from replicated elutions of Tephritidae and Syrphidae of A whole body digestions and B leg digestions per DNA extraction method (Qiagen, DNeasy Blood and Tissue Kit, cat. 69506; QIAamp DNA Mini, cat. 51304; QIAamp DNA Micro, cat. 56304; MinElute PCR Purification, cat. 28004).
Our results show a general trend of decreasing recovery of DNA from older specimens compared to younger specimens (Fig.

Boxplots per collection year for Tephritidae and Syrphidae specimens extracted with the DNeasy Blood and Tissue Kit (Qiagen): A DNA quantities (calculated from concentration measured with Qubit 4.0); B the proportion of DNA fragments between 35 and 350 bp (measured with Fragment Analyzer (DNF-930 dsDNA Reagent kit)); C proportion of sequenced reads with Q > 30.
Sub-optimal or low-quality DNA from museum specimens is often not directly suitable for genetic/genomic analyses (
The DNA of these diverse and heterogeneously collected samples, even if generally suboptimal in terms of concentration, fragmentation and contamination, still allowed recovery of substantial amounts of quality reads (Q > 30) of potential use for genomic research. This general approach needs to be complemented with more specialised and time/cost-demanding procedures for highly-degraded DNA from older specimens. A two-step approach, including the use of commercial kits and methods outlined here allows for rapid screening of younger specimens and reserving the more intensive protocols (also including aDNA methodologies) for older specimens represents a pragmatic, cost-effective route to the routine genotyping of our insect collections.
Main findings and recommendations:
-
The DNeasy Blood and Tissue Kit (Qiagen) provided a cost-effective method of extracting DNA from museum specimens aged one to 25 years;
-
These recently-collected samples, although containing fragmented DNA, represent a tractable tissue source for large-scale sequencing projects;
-
For older material, the use of low input library preparation for highly-fragmented and low concentration DNA extracts is recommended.
Data availability: The data and meta-data from this case study are documented in Suppl. material
Case study 3: Genome skimming as a tool to recover whole plastid genomes from threatened Central African timber species
Introduction
Worldwide, multiple tree species used for timber production are under severe threat (Fig.

The quality and quantity of DNA in herbarium specimens is strongly reliant on collection and storage conditions and, in general, herbarium DNA can be highly fragmented (< 150 bp) and only available in very low amounts (< 5 ng/µl). Interestingly, the techniques optimised for historical herbarium specimens can also be applied to heartwood specimens (i.e. old degenerated material) of processed wood. By jointly analysing herbarium material and silica-dried leaf samples, a clear comparison can be made on the feasibility of historical material for genome skimming purposes, with the aim of yielding full plastid genomes of selected species that are under strong pressure due to illegal logging activities in Central Africa.
Materials and Methods
In order to obtain plastomes of the most important timber species from equatorial African tree species, we collected leaf tissue samples (2 cm2; ca. 10 mg) from 12 herbarium specimens and 20 silica samples via various herbaria (BR, BRLU & L; Suppl. material
DNA extraction and library preparation
Total genomic DNA of both silica and herbarium material was extracted using a combined and modified version of the CTAB protocol (
The purity of the resulting DNA was measured under the absorbance ratios OD 260/280 and OD 260/230 using a NanoDrop 2000 (Thermo Fisher Scientific, US). DNA concentration (ng/μl) and fragment size distribution were measured by capillary electrophoresis using a Fragment Analyzer (Agilent, US). Library preparation of the silica-dried leaf material was initiated via an enzymatic DNA fragmentation step with the aim to retain DNA fragments with a size between 200 and 450 bp after which an end repair step took place. This step was conducted using the NEBnext UltraTM II FS DNA Library Prep Kit for Illumina (New England Biolabs, US). Due to the presence of already degraded DNA in the herbarium specimens, the enzymatic DNA fragmentation step was not carried out for the herbarium material. Adapter ligation was conducted using the NEB-next Adaptor kit for Illumina and U-excision was carried out using the USER Enzyme kit (New England Biolabs, US). Size selection (320–470 bp) was conducted following the SPRIselect protocol (Beckman Coulter, US). With the NEBNext Ultra II Q5 Master Mix, adaptor-ligated DNA was indexed, then PCR-enriched with the NEBNext Multiplex Oligos for Illumina (New England Biolabs, US). For the latter, the following thermocycler reactions were used: Initial denaturation at 98°C for 30 s, 3–4 cycles of denaturation at 98°C, each for 10 s as well as an annealing/extension at 65°C for 75 s and a final extension phase at 65°C for 5 min. In the last step of the library preparation, a DNA-library purification was conducted using SPRIselect (Beckman Coulter, US). The final fragment size distribution and molarity (nM) were examined with a Fragment Analyzer (Agilent, US). Indexed libraries were subsequently pooled (on average 25 samples per lane) in equimolar ratios. Sequencing of the DNA libraries (low coverage paired-end; 10X, 150 bp) was done on a HiSeq 3000, a HiSeq 4000 and NovaSeq 6000 (Illumina, US). At the time of analysis, between autumn 2019 and spring 2021, library preparation and sequencing costs were estimated at a total of €45 - 50 per sample.
Data analysis
The quality of the raw reads was evaluated with FastQC (
Results and Discussion
Amongst the 16 herbarium specimens, DNA yields varied between 220 and 430 ng/µl, whereas DNA yields of silica-dried samples varied between 210 and 850 ng/µl (based on starting leaf tissue samples of 2 cm2). All taxa investigated yielded sufficient DNA and were used for library preparation and sequencing. For the herbarium specimens, between 200,000 and 5.2 million high-quality paired-end reads were produced, whereas for the silica-dried samples, this amount varied between 1.6 million and 6.4 million reads. Over 4 million high-quality paired-end reads were retrieved for only 19% of the herbarium specimens, whereas for the silica-dried samples, there was a ca. 50/50 split of accessions above or below 4 million high-quality paired-end reads. Genome coverage depth was at least 50x for all specimens with fully assembled plastid genomes. Even though a very small number of reads were retrieved from some specimens, it was possible to generate complete plastomes for the majority of the herbarium specimens (
The results obtained in this case study corroborate those of some recently-published studies on the use of genome skimming for the retrieval of full plastomes of land plants (
Main findings and recommendations:
-
Genome skimming of herbarium specimens has shown high success rates across multiple independent studies;
-
Despite the often lower number of reads retrieved from herbarium specimens compared to fresh tissue, it is becoming increasingly routine to generate complete (or almost complete) plastomes from herbarium material using genome skimming;
-
Since one of the most important steps in the genome skimming protocol is to downsize the DNA fragment length, the often highly-degraded DNA of herbarium specimens allows the sonication step to be bypassed in the library preparation protocol.
Data availability: The data in this case study are available under the following GenBank numbers: MZ274087, MZ274092, MZ274094-MZ274096, MZ274099, MZ274102-MZ274107, MZ274110, MZ274113, MZ274116-MZ274122, MZ274124, MZ274127-MZ274129, MZ274132, MZ274135, MZ274137, MZ274143, MZ274145, MZ274147, MZ274148 (see
Case study 4: Comparing hybridisation capture derived sequences from herbarium specimens with data from living material of the same genetic individuals
Introduction
Herbarium collections worldwide contain an estimated 350 million specimens collected over the last approximately 400 years (
The preservation and quality of DNA in herbarium material are highly variable. It has been suggested that DNA decays at a faster rate in plant remains compared to animals (
In this study, we sampled specimens from the herbarium at the Royal Botanic Garden Edinburgh (RBGE) that were collected 12-50 years ago from cultivated individuals of Rhododendron javanicum. These cultivated individuals are still present as live plants in the living collection at RBGE and allowed us to assess the reliability of sequences recovered from herbarium material compared to freshly-collected samples from the same genetic individuals. The chosen sequencing approach was hybridisation capture (also known as target capture or target DNA enrichment) which is an effective sequencing approach for studies utilising degraded DNA sources because it enables recovery of sequence data from low concentrations of endogenous DNA (
Materials and Methods
Samples
Twelve RBGE herbarium vouchers (dated 1972-2010) of various sub-species of cultivated Rhododendron javanicum were sampled along with fresh leaf material from 10 living individuals growing in the RBGE glasshouses, from which the herbarium vouchers were generated. Two of the living individuals were represented by two separate vouchers, collected one or ten years apart. Herbarium samples and their corresponding living samples are listed in Table
Details of Rhododendron herbarium samples used in this study including collection and accession numbers, as well as library protocols used. RHD002 and RHD007 herbarium specimens relate to the same single individual in the living collection, as do RHD016 and RHD018, respectively. Two samples (RHD011 and RHD018) had sequencing libraries prepared using two different protocols. Fresh samples from the living collection were also collected for all individuals. DNA fragment size distribution: size as stated, except bimodal which means one peak of < 1000 bp and one peak of approximately 1-20 kbp. ssDNA = single-stranded DNA library, NEB = NEBNext Ultra II library with sonicated DNA. *All sequencing libraries for the living collection were prepared using NEBNext Ultra II kits with sonicated DNA.
|
Sample |
Species |
Subspecies |
RBGE herbarium collection number |
Specimen date |
RBGE living collection accession number |
DNA fragment size distribution |
Library protocol(s) for herbarium samples* |
|
RHD002 |
Rhododendron javanicum |
kinabaluense |
E00421003 |
2010 |
19801291A |
bimodal |
NEB |
|
RHD003 |
R. javanicum |
moultonii |
E00294943 |
2009 |
20110223A |
bimodal |
NEB |
|
RHD005 |
R. javanicum |
E00328126 |
2009 |
19672627A |
100-1000 bp |
NEB |
|
|
RHD006 |
R. javanicum |
brookeanum |
E00328133 |
2009 |
19801298C |
bimodal |
NEB |
|
RHD007 |
R. javanicum |
kinabaluense |
E00328548 |
2009 |
19801291A |
bimodal |
NEB |
|
RHD008 |
R. javanicum |
palawanense |
E00294512 |
2008 |
19922762B |
bimodal |
NEB |
|
RHD009 |
R. javanicum |
cladotrichum |
E00294755 |
2007 |
19913084A |
bimodal |
NEB |
|
RHD011 |
R. javanicum |
palawanense |
E00954297 |
1998 |
19922772 |
bimodal |
ssDNA, NEB |
|
RHD013 |
R. javanicum |
javanicum |
E00954260 |
1990 |
19730741 |
< 500 bp |
ssDNA |
|
RHD016 |
R. javanicum |
javanicum |
E01016321 |
1982 |
19680840 |
< 500 bp |
ssDNA |
|
RHD017 |
R. javanicum |
kinabaluense |
E01016323 |
1981 |
19690955 |
< 500 bp |
ssDNA |
|
RHD018 |
R. javanicum |
javanicum |
E01016322 |
1972 |
19680840 |
< 500 bp (+tail) |
ssDNA, NEB |
DNA extraction and library preparation - herbarium samples
DNA from herbarium specimens was extracted as described in
DNA extracts with fragments shorter than 500 bp (n = 5) were converted into single-stranded DNA (ssDNA) libraries following
Of the remaining samples (n = 9), we recovered less-fragmented DNA, including samples with a bimodal DNA fragment size distribution (n = 7), with one peak of fragments shorter than 1000 bp and a second peak of approximately 1-20 kbp; the other samples included one sample (RHD005) with DNA fragments of 100-1000 bp and one (RHD018) with mostly short fragments, but with a tail of longer fragments. Aliquots of these extracts were subjected to 8-12 sonication cycles of 30 s on, 90 s off, using a Diagenode Bioruptor sonicator, for a target fragment size of 200-400 bp. Libraries were generated with the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina (New England Biolabs) and indexed with NEBNext® Multiplex Oligos for Illumina® (Unique Dual Index Primer Pairs). Because these libraries were produced in non-dedicated facilities where fresh plant material is regularly processed — posing a risk for contamination — the following anti-contamination precautions were taken: pre-amplification steps were carried out inside a dedicated laminar flow hood in a pre-PCR room with dedicated reagents and consumables and negative non-template controls were included.
DNA extraction and library preparation - living collection samples
Approximately 150 mg of leaf material was harvested into 7.6 ml FluidX tubes and placed immediately into liquid nitrogen. DNA was extracted using a protocol developed for extracting high molecular weight DNA (
Hybridisation capture and sequencing
Hybridisation capture was performed on all libraries. The assay was designed using two published Rhododendron genomes from NCBI: R. delavayi (
Libraries were pooled according to material and library construction protocol. The samples were processed with a wider set of samples than are presented here, such that each pool contained 10-14 libraries. Negative controls were pooled separately. Hybridisation capture was performed following the MyBaits (Arbor Biosciences) protocol v.5.02 with the high sensitivity version and the hybridisation and wash temperatures set to 63°C for herbarium samples (the second round of enrichment was omitted) and with the standard version and hybridisation and wash temperatures set to 65°C for living samples. Pools were re-amplified post-capture in two 50 μl reactions with Herculase II Fusion DNA polymerase (Agilent) for 14 cycles. Captured libraries for living and herbarium samples were sequenced on separate Illumina MiSeq lanes with no index repetition, with 150 bp PE v.2 runs (4.5 - 5.0 Gb) at the University of Exeter sequencing facilities. The target region of the bait set is 621,078 bp, which translates to an average coverage of 720X/sample for the 10 living samples and 600X for the 12 herbarium samples.
Data analysis
Herbarium reads were processed with the PALEOMIX v.1.3.7 BAM pipeline (
Quantity and quality of the SNPs called for the herbarium samples were assessed by comparison to the sequence from their respective paired living sample. First, a new reference for each individual was generated using sequence data from only the living sample of that individual. BAM files from the initial run of PALEOMIX (above) were filtered using strict settings on bcftools v.1.16 (filter SNPs by QUAL > 160 and DP > 10) and consensus fasta files were generated to be used as a new reference (
Results and Discussion
Evaluating sequencing library preparation for herbarium material and contamination control
Without any prior assumption of DNA fragmentation rates in the herbarium samples processed in this study, our approach consisted of isolating DNA in dedicated clean facilities. Following an assessment of DNA fragment size, we decided to separate samples into two categories. Fragmented DNA extracts were treated, following aDNA protocols in dedicated facilities, whereas extracts containing longer DNA fragments were taken to non-dedicated facilities for DNA shearing and library preparation using commercially available kits. We assessed coverage of targeted loci (Fig.

Sequencing coverage of targeted loci and library complexity for Rhododendron specimens. A Coverage of targeted nuclear loci. B Proportion of PCR duplicates. LIV = libraries generated from living collection samples, ssDNA = single-stranded DNA libraries made from degraded herbarium DNA, NEB = double-stranded DNA libraries made from sheared herbarium DNA using a commercial kit. C DNA deamination patterns of read data obtained from NEB (red, blue) and ssDNA (dark red, dark blue) herbarium libraries with mapDamage v.2.2.1. First base was removed for visualisation.
We did not detect any amplification products in the negative controls of libraries generated in the non-dedicated facilities following anti-contamination measures, indicating that it was possible to process herbarium DNA extracts of sufficient DNA concentration and fragment size under these conditions and with the necessary precautions. In addition to the ssDNA library construction protocol (
Finally, we observed mild deamination patterns in reads recovered from herbarium material (Fig.
Assessing reliability of SNPs recovered from herbarium material
We took advantage of cultivated plants present in the RBGE living collection, from which herbarium vouchers were created 12-50 years ago, to investigate whether sequences recovered from the herbarium samples were an accurate biological replicate of the living material, or if low starting templates and base modifications, both features that accumulate in degrading DNA over time, resulted in erroneously-called bases (
We typically observed 75-108 SNPs per individual, of which 45-87 were shared between living and herbarium samples (Fig.

Comparison of SNPs recovered from herbarium and living collection samples of the same individuals of Rhododendron species. A Number of SNPs called, categorised into exclusive to living samples (light blue, likely caused by ambiguous calls at heterozygous sites), exclusive to herbarium samples (dark blue, likely caused by sequencing errors due to degraded DNA) or shared (yellow). B Depth and C quality of shared and herbarium-exclusive SNPs. ssDNA = single-stranded DNA libraries made from degraded herbarium DNA, NEB = double-stranded DNA libraries made from sheared herbarium DNA using a commercial kit.
Two samples (represented by three libraries) showed a noticeable spike in SNP abundance compared to all others. Both of these samples are from the same sub-species Rhododendron javanicum ssp. palawanense (RHD008 and RHD011) and retrospective analyses of their morphology suggest they may be of hybrid origin. It is possible that the observed spike in SNP abundance is due to these specimens having higher levels of heterozygosity due to hybridity. With a greater number of variable sites, there is an associated increased possibility of detecting both genuine (shared) SNPs with respect to the reference, as well as a corresponding increase in erroneous SNPs due to poor coverage of these sites in herbarium material.
Implications for sequencing herbarium specimens
Multiple studies have now been undertaken exploring the potential of hybridisation capture for the recovery of sequence data from herbarium specimens. These have included exploratory studies assessing the feasibility of the approach for recovering sequence data from plant specimens with a range of different ages (
Main findings and r ecommendations:
-
Hybridisation capture is now well established as a method for recovery of large amounts of nuclear sequence data from herbarium specimens and the approach works well in accommodating the complexity of plant genomes;
-
Studies recovering DNA from herbarium specimens should take place in dedicated clean or low-copy facilities. Once DNA fragment length distribution is known, sequencing library preparation can take place according to DNA size;
-
Library preparation from highly-fragmented DNA should take place in dedicated clean or low-copy facilities. We found the ssDNA protocol by
Kapp et al. (2021) to be fast and efficient for this purpose in the current study and the simplicity of this approach warrants further trialling to see if these results are generally applicable; -
DNA extracts that show a bimodal fragment size distribution with the majority of fragments > 1 kb can be sheared, prior to library preparation with a commercially-available kit. If this takes place in non-dedicated facilities, we recommend the following contamination-limiting precautions:
-
Physical separation of pre- and post-PCR laboratories;
-
Dedicated laminar flow hood for all pre-PCR steps (to be regularly decontaminated);
-
Dedicated reagents and consumables;
-
Inclusion of non-template negative controls;
-
-
Distribution of SNP quality and coverage should be inspected for a better-informed decision on filtering parameters. Stringent quality filtering of SNPs can provide high confidence in genotype calls even from herbarium material.
Data availability:
The raw sequencing data has been deposited at the European Nucleotide Archive (ENA) under project PRJEB61704 with accession IDs ERS15567903 - ERS15567924.
Case study 5: Selecting samples with the greatest likelihood of success for reduced-representation sequencing from museum collections
Introduction
Reduced-representation sequencing (RRS) using restriction digests followed by fragment sequencing is a cost-effective route for generating thousands of genetic markers (
Materials and Methods
Sampling
We sampled 96 barn owls (Tyto alba alba) comprising both historical as well as contemporary specimens. Historical samples were obtained from collections stored at the Royal Belgian Institute of Natural Sciences and covered two distinct periods in time, mainly from the 1930s (1929-1943, n = 15) and mainly from the 1970s (1966-1979, n = 22). Contemporary specimens (n = 59) comprised road kills which were brought to bird sanctuaries and stored in freezers immediately upon arrival. We collected toe pads of all historical specimens to minimise voucher damage and liver or breast muscle tissue of the contemporary specimens.
DNA extraction, library preparation and SNP calling
DNA of all specimens was extracted using the NucleoSpin Tissue Kit (Macherey-Nagel GmbH). Concentrations were quantified by the Qubit fluorometer (Invitrogen) and a fragment analysis of historical samples was conducted on a 2100 Bioanalyzer (Agilent). While numerous variations on reduced-representation genome sequencing exist (
Contamination assessment
In order to avoid any bias in downstream analyses arising from contaminated historical specimens, we first assembled a stringently-filtered vcf based exclusively on recent samples. Specimens showing more than 20% missing data were discarded and only biallelic SNPs (--max-alleles 2) with a minimal SNP quality (--minQ) of 40 and an individual genotype (--minGQ) quality of 30, present in at least 50% of the individuals (--max-missing) and a minimum allele frequency (--maf) of 0.01 were retained with VCFtools (
Statistical analysis
We ran a one-way ANOVA to test for differences in mean number of missing SNPs amongst the three time periods and allowed for period-specific variances to account for heteroscedasticity using the R package ‘nlme’ (
Results and Discussion
Mean missing data per individual differed significantly between time periods (chi-square = 62.56, p < 0.001) (Fig.

Mean DNA concentration in historical and recent samples were respectively 20.2 ng/µl ± 12.4 (SD) and 30.6 ng/µl ± 13.9 (SD). A simple linear regression indicated the number of missing SNPs was not related to DNA concentration in recent samples (F1,57 = 0.016, p = 0.90). In contrast, a GAM indicated DNA concentration was inversely related to the amount of missing data in historical samples (F1,3.2 = 15.97, p < 0.001) and explained 66.8% of the deviance (Fig.

GAMs relating the amount of missing data to the percentage of fragments between 35-200 bp, 200-400 bp, 400-700 bp and 700-10,380 bp explained, respectively, 74.8%, 20.7%, 39.7% and 78.4% of the model deviance. The amount of fragments in the lowest bin range was strongly positively associated with the levels of missing data (F1,2.3 = 32.99, p < 0.001), while those at the highest bin range showed a clear negative association (F1,2.4 = 37.63, p < 0.001) (Fig.

To date, few studies have assessed whether RRS on museum collections is feasible, and if so, how to optimise approaches. In a previous study using ddRAD target enriched sequencing, an inclusion threshold for DNA concentration of 30 ng/µl was suggested (as determined from the A260 values) (
ddRAD appears unsuitable to obtain sequence data from highly-fragmented samples (in our case, the older museum samples dating from the 1930s and some more recently collected material from the 1970s). More advanced target-capture-based technologies such as HyRAD and HyRAD-X should be considered as an alternative (
Main findings and recommendations :
-
ddRAD cannot be routinely applied to large museum collections to obtain population-level genomic data, especially when dealing with heavily-fragmented samples;
-
However, despite the challenges of using ddRAD on degraded DNA, we were able to obtain ddRAD seq data from avian samples up to ca. 50 years old, and screening the fragment profiles of the genomic DNA gave good predictions of levels of missing data;
-
Such screening is relatively easy to accomplish at minimal cost by any moderately-equipped molecular lab and substantially reduces the risk of both data loss and unnecessary library preparation and sequencing costs;
-
The inclusion of data from high-quality fresh samples is important to establish a reference set to aid targeting endogenous sequence data from museum specimens.
Data availability: The raw sequencing data from this case study have been deposited at the European Nucleotide Archive (ENA) under project PRJEB59169 with accession IDs ERS14470037 - ERS14470133.
Case study 6: Single-tube DNA library preparation for ancient bones
Introduction
Massive parallel sequencing based on sequencing-by-synthesis technologies (Illumina) is an efficient method for generating DNA data from ancient material because it recovers sequences from large amounts of very short DNA fragments. In preparing samples for sequencing, single-tube DNA library protocols circumvent inter-reaction purification steps which require the transfer of DNA solutions to new tubes. They were shown to reduce DNA loss, preparation time and expenses compared to other DNA library preparation methods (
Materials and Methods
We selected seven bones of Bovidae of different ages (Table
Bovid bone sample information, isolated DNA concentrations and proportions of reads mapped to target and possible contaminant full genomes. ID: Tissue sample identification; conc: DNA concentration in the DNA extract measured using Qubit; mapped: percentages of the deduplicated paired-end reads mapping to the reference genomes of Bos taurus, Homo sapiens and Mus musculus (separated by “/”); short: percentages of mapped reads smaller than 100 bp; long: percentages of mapped reads longer than 300 bp (with insert between the paired reads); Neg1 and Neg2: negative DNA extracts processed for each library.
|
ID |
Epoch |
Conc |
Mapped |
Short |
Long |
|
ng/µl |
% N raw |
% < 100 bp |
% > 300 bp |
||
|
LAST1 |
Roman period |
1.4 |
0.157/0.06/0.077 |
83.43/2.93/1.79 |
0.52/10.05/10.58 |
|
LAST2 |
Roman period |
11.8 |
0.644/0.01/0.004 |
92.87/36.08/39.93 |
0.31/17.67/7.72 |
|
LAST3 |
Roman period |
2.6 |
1.674/0.055/0.073 |
85.84/14.15/5.65 |
0.42/6.14/9.98 |
|
LAST4 |
Roman period |
3.7 |
0.02/0.03/0.054 |
72.39/3.7/1.88 |
1.38/7.71/10.1 |
|
LAST5 |
Roman period |
5.2 |
0.123/0.017/0.005 |
88.27/11.29/19.94 |
0.39/26.95/9.76 |
|
LAST7 |
epipaleolithic |
7.8 |
0.033/0.01/0.003 |
98.01/13.29/21.84 |
0.49/24.7/10.18 |
|
LAST9 |
late medieval |
0.5 |
7.844/0.179/0.169 |
84.91/16.83/6.91 |
0.33/8.08/11.46 |
|
Neg1 |
NA |
0 |
0.058/1.55/0.417 |
59.44/3.18/0.31 |
3.89/29.07/13.07 |
|
Neg3 |
NA |
0 |
0.278/2.828/2.382 |
18.01/1.53/0.92 |
5.64/2.47/12.43 |
A total of 7 to 51 ng of genomic DNA of each specimen was used as starting material for the ‘Ultra’ single-tube DNA library preparation method described in
In total, 225.52 million reads (33.828 Gbp) were generated for the seven specimens and the two controls (Suppl. material
Results and Discussion
DNA concentrations after DNA extraction ranged from 0 to 11.8 ng/µl (Table
Data authentication
The single-tube library preparation protocol applied here (
Main findings and recommendations:
-
Streamlined single-tube DNA library preparation methods adapted for degraded DNA and followed by shallow shotgun sequencing are useful for estimating the percentages of ancient endogenous DNA recoverable from DNA extracts;
-
Data authentication should integrate as many aspects of the endogenous DNA as possible. Comparisons with controls (extraction negatives and unrelated genomic data) are crucial to assess the risk of including contaminant DNA in the analysis and to guide steps to filter them out;
-
DNA fragment-size profiles of the DNA extracts are indicative of the presence of degraded DNA, but sequencing is necessary to evaluate percentages of endogenous DNA.
Data availability: The raw sequencing data for this case study have been deposited at the European Nucleotide Archive (ENA) under project PRJEB59185 with accession IDs ERS14471070 - ERS14471078.
Concluding remarks
The continually evolving landscape of sequencing platforms and chemistries is resulting in an ever-expanding set of opportunities for unlocking the genomic resources held in natural history collections and there is a general increase in the feasibility of museum specimen sequencing. With the rapid expansion in the field of museomics, comes a pressing need for the ongoing development, sharing and adoption of best practices. Areas of particular importance include establishment of appropriate facilities, workflows and data verification steps to minimise risks of contamination and sampling guidance which supports optimal utilisation of museum specimens for genomic research. Another area of general importance is attention to ethical issues associated with the use of specimens for genomic science; many collections pre-date contemporary permit conditions or restrictions. Guidelines for ethical issues associated with sampling specimens for genomic analysis are mostly developed for human tissues and archaeofaunal remains (
Acknowledgements
We are grateful to Sean Prosser and Evegeny Zakharov (University of Guelph), Ben Price (Natural History Museum London), Ryan Folk (Mississippi State University) and Chris Raxworthy (American Museum of Natural History) for advice; to five reviewers and the subject editor for their comments on the paper; to Suzanne Cubey at the Royal Botanic Garden Edinburgh for facilitating access to the herbarium collection for tissue sampling; to the Department of Biosciences at the University of Oslo for access to the ancient DNA laboratory; to Bea De Cupere, Quentin Goffette, Mietje Germonpré and Annelise Folie from the Royal Belgian Institute of Natural Sciences, Belgium for designing the project “LAST”, authorising tissue sampling, giving access to RBINS specimens and to their associated information; to the Kerkuilenwerkgroep Vlaanderen for providing tissue samples; to Lejia Zhang (MfN Berlin) for isolating DNA and preparing libraries and Susan Mbedi and Sarah Sparman (Berlin Center for Genomics in Biodiversity Research) for conducting quality checks and sequencing.
Funding program
This research received support from the SYNTHESYS+ Project (http://www.synthesys.info/) which was financed by European Community Research Infrastructure Action under the H2020 Integrating Activities Programme, Project number 823827. The Royal Botanic Garden Edinburgh acknowledges support from the Scottish Government's Rural and Environment Science and Analytical Services Division (RESAS). Analyses at RBGE were run on the Crop Diversity Bioinformatics Resource which is funded by BBSRC BB/S019669/1. The Joint Experimental Molecular Unit (JEMU) of the Royal Belgian Institute of Natural Sciences (RBINS, Brussels) and the Royal Museum for Central Africa (RMCA, Tervuren) acknowledge funding support from the Belgian Science Policy (Belspo).
Grant title
This work was funded as part of the SYNTHESYS+ Project (http://www.synthesys.info)
Author contributions
GF, LE, MLH, SJ, TvR, GS, CV, MV and PMH conceived and designed the study, GF and PMH produced the initial draft of paper and all authors contributed to augmenting, refining and revising the manuscript. Case study 1 (insect mitogenomics) was led by TvR and JP; Case study 2 (WGS of insects) by LE and MV; Case study 3 (genome skimming of plants) by SJ and MM; Case study 4 (target capture of plants) by GF, MLH, CK, FP and PMH; Case study 5 (RAD seq of birds) by CV; Case study 6 (bovid bone sequencing) by GS.
Conflicts of interest
References
- Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries.Genome Biology12(2):R18. https://doi.org/10.1186/gb-2011-12-2-r18
- The half-life of DNA in bone: measuring decay kinetics in 158 dated fossils.Proceedings of the Royal Society: Biological Sciences279(1748):4724‑4733. https://doi.org/10.1098/rspb.2012.1745
- The treasure vault can be opened: large-scale genome skimming works well using herbarium and silica gel dried aterial.Plants9(4):432. https://doi.org/10.3390/plants9040432
- Methodologies for ancient DNA extraction from bones for genomic analysis: Approaches and guidelines.Russian Journal of Genetics58(9):1017‑1035. https://doi.org/10.1134/s1022795422090034
- FastQC A quality control tool for high throughput sequence data (Online).Available online at:URL: https://www.bioinformatics.babraham.ac.uk/projects/fastqc
- To curate the molecular past, museums need a carefully considered set of best practices.Proceedings of the National Academy of Sciences of the United States of America116(5):1471‑1474. https://doi.org/10.1073/pnas.1822038116
- Herbarium genomics: plastome sequence assembly from a range of herbarium specimens using an Iterative Organelle Genome Assembly pipeline.Biological Journal of the Linnean Society117(1):33‑43. https://doi.org/10.1111/bij.12642
- SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing.Journal of Computational Biology19(5):455‑477. https://doi.org/10.1089/cmb.2012.0021
- Herbaria are a major frontier for species discovery.Proceedings of the National Academy of Sciences of the United States of America107(51):22169‑22171. https://doi.org/10.1073/pnas.1011841108
- Valuing museum specimens: high-throughput DNA sequencing on historical collections of New Guinea crowned pigeons (Goura).Biological Journal of the Linnean Society117(1):71‑82. https://doi.org/10.1111/bij.12494
- Herbarium-based science in the twenty-first century.Botany Letters165(3-4):323‑327. https://doi.org/10.1080/23818107.2018.1482783
- Metagenomic analysis of historical herbarium specimens reveals a postmortem microbial community.Molecular Ecology Resources20(5):1206‑1219. https://doi.org/10.1111/1755-0998.13174
- Historical DNA as a tool to address key questions in avian biology and evolution: A review of methods, challenges, applications, and future directions.Molecular Ecology Resources19(5):1115‑1130. https://doi.org/10.1111/1755-0998.13066
- Sequence capture and phylogenetic utility of genomic ultraconserved elements obtained from pinned insect Specimens.PLOS One11(8):e0161531. https://doi.org/10.1371/journal.pone.0161531
- Trimmomatic: a flexible trimmer for Illumina sequence data.Bioinformatics30(15):2114‑2120. https://doi.org/10.1093/bioinformatics/btu170
- Genetic time traveling: sequencing old herbarium specimens, including the oldest herbarium specimen sequenced from kingdom Fungi, reveals the population structure of an agriculturally significant rust.New Phytologist237(4):1463‑1473. https://doi.org/10.1111/nph.18622
- Factors affecting targeted sequencing of 353 nuclear genes from herbarium specimens spanning the diversity of angiosperms.Frontiers in Plant Science10:1102. https://doi.org/10.3389/fpls.2019.01102
- Patterns of damage in genomic DNA sequences from a Neandertal.Proceedings of the National Academy of Sciences USA37:14616‑14621. https://doi.org/10.1073/pnas.0704665104
- Novel high-resolution characterization of ancient DNA reveals C > U-type base modification events as the sole cause of post mortem miscoding lesions.Nucleic Acids Research35(17):5717‑5728. https://doi.org/10.1093/nar/gkm588
- Properties of formaldehyde-treated nucleohistone.Biochemistry8(8):3214‑3218. https://doi.org/10.1021/bi00836a013
- The use of museum specimens with high-throughput DNA sequencers.Journal of Human Evolution79:35‑44. https://doi.org/10.1016/j.jhevol.2014.10.015
- BLAST+: architecture and applications.BMC Bioinformatics10:421. https://doi.org/10.1186/1471-2105-10-421
- An accounting approach to calculate the financial value of a natural history collection of mammals in Ecuador.Museum Management and Curatorship33(3):279‑296. https://doi.org/10.1080/09647775.2018.1466191
- DNA from museum collections. In:Molecular identification of plants: from sequence to species.Vol. 1.Advanced Bookshttps://doi.org/10.3897/ab.e98875
- Museum genomics.Annual Review of Genetics55:633‑659. https://doi.org/10.1146/annurev-genet-071719-020506
- Single-tube library preparation for degraded DNA.Methods in Ecology and Evolution9(2):410‑419. https://doi.org/10.1111/2041-210X.12871
- Pulling out the 1%: whole-genome capture for the targeted enrichment of ancient DNA sequencing libraries.American Journal of Human Genetics93(5):852‑864. https://doi.org/10.1016/j.ajhg.2013.10.002
- Care and Conservation of Natural History Collections.Oxford: Butterworth HeinemannURL: http://www.natsca.org/care-and-conservation
- A method for counting PCR template molecules with application to next-generation sequencing.Nucleic Acids Research39(12):e81. https://doi.org/10.1093/nar/gkr217
- Stacks: building and genotyping loci de novo from short-read sequences.G3, Genes, Genomes, Genetics1(3):171‑182. https://doi.org/10.1534/g3.111.000240
- To bee, or not to bee? One leg is the question.Molecular Ecology Resources22(5):1868‑1874. https://doi.org/10.1111/1755-0998.13578
- Poor hDNA-derived NGS data may provide sufficient phylogenetic information of potentially extinct taxa.Frontiers in Ecology and Evolution10:907889. https://doi.org/10.3389/fevo.2022.907889
- A linked-read approach to museomics: Higher quality de novo genome assemblies from degraded tissues.Molecular Ecology Resources20(4):856‑870. https://doi.org/10.1111/1755-0998.13155
- Genomics reveals the origins of historical specimens.Molecular Biology and Evolution38(5):2166‑2176. https://doi.org/10.1093/molbev/msab013
- CETAF Code of Conduct and Best Practice. .
- Mitochondrial metagenomics: letting the genes out of the bottle.GigaScience5(1):s13742-016-0120-y. https://doi.org/10.1186/s13742-016-0120-y
- Genome sequencing of museum specimens reveals rapid changes in the genetic composition of honey bees in California.Genome Biology and Evolution10(2):458‑472. https://doi.org/10.1093/gbe/evy007
- Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology.Nucleic Acids Research36(19):e122. https://doi.org/10.1093/nar/gkn502
- Length and GC-biases during sequencing library amplification: a comparison of various polymerase-buffer systems with ancient and modern DNA sequencing libraries.BioTechniques52(2):87‑94. https://doi.org/10.2144/000113809
- Ancient DNA damage.Cold Spring Harbor Perspectives in Biology5(7):a012567. https://doi.org/10.1101/cshperspect.a012567
- Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments.Proceedings of the National Academy of Sciences of the United States of America110(39):15758‑15763. https://doi.org/10.1073/pnas.1314445110
- Modeling genome coverage in single-cell sequencing.Bioinformatics30(22):3159‑3165. https://doi.org/10.1093/bioinformatics/btu540
- The variant call format and VCFtools.Bioinformatics27(15):2156‑2158. https://doi.org/10.1093/bioinformatics/btr330
- RADSeq: next-generation population genetics.Briefings in Functional Genomics9(5-6):416‑423. https://doi.org/10.1093/bfgp/elq031
- Genome-wide genetic marker discovery and genotyping using next-generation sequencing.Nature Reviews Genetics12(7):499‑510. https://doi.org/10.1038/nrg3012
- Quantifying and reducing spurious alignments for the analysis of ultra-short ancient DNA sequences.BMC Biology16:121. https://doi.org/10.1186/s12915-018-0581-9
- Policies Handbook on Using Molecular Collections.ARPHA Preprintshttps://doi.org/10.3897/arphapreprints.e98432
- Tales from the crypt: genome mining from fungarium specimens improves resolution of the mushroom tree of life.Biological Journal of the Linnean Society117(1):11‑32. https://doi.org/10.1111/bij.12553
- A SMRT approach for targeted amplicon sequencing of museum specimens (Lepidoptera)-patterns of nucleotide misincorporation.PeerJ9https://doi.org/10.7717/peerj.10420
- A rapid DNA isolation procedure for small quantities of fresh leaf tissue.Phytochemical Bulletin19:11‑15.
- Dichlorvos exposure impedes extraction and amplification of DNA from insects in museum collections.Frontiers in Zoology7:2. https://doi.org/10.1186/1742-9994-7-2
- Museum specimens provide reliable SNP data for population genomic analysis of a widely distributed but threatened cockatoo species.Molecular Ecology Resources19(6):1578‑1592. https://doi.org/10.1111/1755-0998.13082
- Variola virus genome sequenced from an eighteenth-century museum specimen supports the recent origin of smallpox.Philosophical Transactions of the Royal Society B375(1812):20190572. https://doi.org/10.1098/rstb.2019.0572
- Competitive mapping allows for the identification and exclusion of human DNA contamination in ancient faunal genomic datasets.BMC Genomics21:844. https://doi.org/10.1186/s12864-020-07229-y
- High-throughput methods for efficiently building massive phylogenies from natural history collections.Applications in Plant Sciences9(2):e11410. https://doi.org/10.1002/aps3.11410
- The Limits of Hyb-Seq for herbarium specimens: impact of preservation techniques.Frontiers in Ecology and Evolution7:439. https://doi.org/10.3389/fevo.2019.00439
- Destructive sampling natural science collections: An overview for museum professionals and researchers.Journal of Natural Science Collections5:21‑34. URL: https://www.natsca.org/sites/default/files/publications/JoNSC-Vol5-FreedmanVanDorpBrace2018.pdf
- Setting Up an Ancient DNA Laboratory. In:Ancient DNA: Methods and Protocols.Humana Press,Totowa, NJ,10pp. [ISBN9781617795169]. https://doi.org/10.1007/978-1-61779-516-9_1
- Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers.BMC Genomics19:531. https://doi.org/10.1186/s12864-018-4933-1
- Assessing ancient DNA studies.Trends in Ecology & Evolution20(10):541‑544. https://doi.org/10.1016/j.tree.2005.07.005
- DNA extraction from dry museum beetles without conferring external morphological damage.PLoS ONE2(3):e272. https://doi.org/10.1371/journal.pone.0000272
- The isolation of nucleic acids from fixed, paraffin-embedded tissues–Which methods are useful when?PLoS ONE2(6):e537. https://doi.org/10.1371/journal.pone.0000537
- Impacts of degraded DNA on restriction enzyme associated DNA sequencing (RADSeq).Molecular Ecology Resources15(6):1304‑1315. https://doi.org/10.1111/1755-0998.12404
- The Neandertal genome and ancient DNA authenticity.The EMBO Journal28(17):2494‑2502. https://doi.org/10.1038/emboj.2009.222
- The origins and adaptation of European potatoes reconstructed from historical genomes.Nature Ecology and Evolution3(7):1093‑1101. https://doi.org/10.1038/s41559-019-0921-3
- Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads - a baiting and iterative mapping approach.Nucleic Acids Research41(13):e129. https://doi.org/10.1093/nar/gkt371
- Unlocking inaccessible historical genomes preserved in formalin.Molecular Ecology Resources22(6):2130‑2147. https://doi.org/10.1111/1755-0998.13505
- Retrieval of hundreds of nuclear loci from herbarium specimens.Taxon65(5):1081‑1092. https://doi.org/10.12705/655.9
- A DNA 'barcode blitz': rapid digitization and sequencing of a natural history collection.PLOS One8(7):e68535. https://doi.org/10.1371/journal.pone.0068535
- Characterizing DNA preservation in degraded specimens of Amara alpina (Carabidae: Coleoptera).Molecular Ecology Resources14(3):606‑615. https://doi.org/10.1111/1755-0998.12205
- BIOSCAN: DNA barcoding to accelerate taxonomy and biogeography for conservation and sustainability.Genome64(3):161‑164. https://doi.org/10.1139/gen-2020-0009
- The use of alcohol in plant collecting.Rhodora49(584):207‑210. URL: http://www.jstor.org/stable/23303840
- DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA.Nucleic Acids Research29(23):4793‑4799. https://doi.org/10.1093/nar/29.23.4793
- Natural history collections as windows on evolutionary processes.Molecular Ecology25(4):864‑881. https://doi.org/10.1111/mec.13529
- A guide to avian museomics: Insights gained from resequencing hundreds of avian study skins.Molecular Ecology Resources22(7):2672‑2684. https://doi.org/10.1111/1755-0998.13660
- Ancient and historical DNA in conservation policy.Trends in Ecology & Evolution37(5):420‑429. https://doi.org/10.1016/j.tree.2021.12.010
- GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes.Genome Biology21:241. https://doi.org/10.1186/s13059-020-02154-5
- Museomics reveals extensive cryptic diversity of Australian prionine longhorn beetles with implications for their classification and conservation.Systematic Entomology45(4):745‑770. https://doi.org/10.1111/syen.12424
- A global approach for natural history museum collections.Science379(6638):1192‑1194. https://doi.org/10.1126/science.adf6434
- A universal probe set for targeted sequencing of 353 nuclear genes from any flowering plant designed using k-medoids clustering.Systematic Biology68(4):594‑606. https://doi.org/10.1093/sysbio/syy086
- mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters.Bioinformatics29(13):1682‑1684. https://doi.org/10.1093/bioinformatics/btt193
- A fast and efficient single-stranded genomic library preparation method optimized for Ancient DNA.Journal of Heredity112(3):241‑249. https://doi.org/10.1093/jhered/esab012
- The effects of herbarium specimen characteristics on short-read NGS sequencing success in nearly 8000 specimens: old, degraded samples have lower DNA yields but consistent sequencing success.Frontiers in Plant Science12:669064. https://doi.org/10.3389/fpls.2021.669064
- MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform.Nucleic Acids Research30(14):3059‑3066. https://doi.org/10.1093/nar/gkf436
- Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data.Bioinformatics28(12):1647‑1649. https://doi.org/10.1093/bioinformatics/bts199
- Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform.Nucleic Acids Research40(1):e3. https://doi.org/10.1093/nar/gkr771
- A new model for ancient DNA decay based on paleogenomic meta-analysis.Nucleic Acids Research45(11):6310‑6320. https://doi.org/10.1093/nar/gkx361
- A 2-million-year-old ecosystem in Greenland uncovered by environmental DNA.Nature612(7939):283‑291. https://doi.org/10.1038/s41586-022-05453-y
- Setting the stage - building and working in an ancient DNA laboratory.Annals of Anatomy194(1):3‑6. https://doi.org/10.1016/j.aanat.2011.03.008
- Cost‐efficient high throughput capture of museum arthropod specimen DNA using PCR‐generated baits.Methods in Ecology and Evolution10(6):841‑852. https://doi.org/10.1111/2041-210x.13169
- A minimally morphologically destructive approach for DNA retrieval and whole genome shotgun sequencing of pinned historic Dipteran vector species.Genome Biology and Evolution13(10):evab226. https://doi.org/10.1093/gbe/evab226
- A complete mtDNA genome of an early modern human from Kostenki, Russia.Current Biology20(3):231‑236. https://doi.org/10.1016/j.cub.2009.11.068
- SNP ascertainment bias in population genetic analyses: why it is important, and how to correct it.Bioessays35(9):780‑786. https://doi.org/10.1002/bies.201300014
- Museomics.Current Biology32(21):R1214‑R1215. https://doi.org/10.1016/j.cub.2022.09.019
- Fast gapped-read alignment with Bowtie 2.Nature Methods9(4):357‑359. https://doi.org/10.1038/nmeth.1923
- Hybridization ddRAD-sequencing for population genomics of nonmodel plants using highly degraded historical specimen DNA.Molecular Ecology Resources20(5):1228‑1247. https://doi.org/10.1111/1755-0998.13168
- Isolation, library preparation, and bioinformatic analysis of historical and ancient plant DNA.Current Protocols in Plant Biology5:e20121. https://doi.org/10.1002/cppb.20121
- A workflow for expanding DNA barcode reference libraries through ‘museum harvesting’ of natural history collections.Biodiversity Data Journal11:e100677. https://doi.org/10.3897/BDJ.11.e100677
- The Earth BioGenome Project 2020: Starting the clock.Proceedings of the National Academy of Sciences of the United States of America119(4):e2115635118. https://doi.org/10.1073/pnas.2115635118
- Fast and accurate short read alignment with Burrows–Wheeler transform.Bioinformatics25(14):1754‑1760. https://doi.org/10.1093/bioinformatics/btp324
- The sequence alignment/map format and SAMtools.Bioinformatics25(16):2078‑2079. https://doi.org/10.1093/bioinformatics/btp352
- Plastid phylogenomic insights into relationships of all flowering plant families.BMC Biology19:232. https://doi.org/10.1186/s12915-021-01166-2
- Instability and decay of the primary structure of DNA.Nature362(6422):709‑715. https://doi.org/10.1038/362709a0
- AdapterRemoval: easy cleaning of next-generation sequencing reads.BMC Research Notes5:337. https://doi.org/10.1186/1756-0500-5-337
- From the field to the laboratory: Controlling DNA contamination in human ancient DNA research in the high-throughput sequencing era.STAR: Science & Technology of Archaeological Research3(1):1‑14. https://doi.org/10.1080/20548923.2016.1258824
- Museomics clarifies the classification of Aloidendron (Asphodelaceae), the iconic African tree Aloes.Frontiers in Plant Science10:1227. https://doi.org/10.3389/fpls.2019.01227
- How to deduplicate PCR.Nature Methods14(5):473‑476. https://doi.org/10.1038/nmeth.4268
- Genome skimming reveals novel plastid markers for the molecular identification of illegally logged African timber species.PLOS One16(6):e0251655. https://doi.org/10.1371/journal.pone.0251655
- Data access for the 1,000 Plants (1KP) project.GigaScience3(1):2047-217X-3-17. https://doi.org/10.1186/2047-217X-3-17
- Adding leaves to the Lepidoptera tree: capturing hundreds of nuclear genes from old museum specimens.Systematic Entomology46(3):649‑671. https://doi.org/10.1111/syen.12481
- Sequence capture of ultraconserved elements from bird museum specimens.Molecular Ecology Resources16(5):1189‑1203. https://doi.org/10.1111/1755-0998.12466
- Performance of commonly requested destructive museum samples for mammalian genomic studies.Journal of Mammalogy99(4):789‑802. https://doi.org/10.1093/jmammal/gyy080
- The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data.Genome Research20(9):1297‑1303. https://doi.org/10.1101/gr.107524.110
- Illumina sequencing library preparation for highly multiplexed target capture and sequencing.Cold Spring Harbor Protocols2010(6):pdb‑prot5448. https://doi.org/10.1101/pdb.prot5448
- Museum genomics confirms that the Lord Howe Island stick insect survived extinction.Current Biology27(20):3157‑3161. https://doi.org/10.1016/j.cub.2017.08.058
- Building natural history collections for the twenty-first century and beyond.BioScience70(8):674‑687. https://doi.org/10.1093/biosci/biaa069
- First large‐scale quantification study of DNA preservation in insects from natural history collections using genome‐wide sequencing.Methods in Ecology and Evolution14(2):360‑371. https://doi.org/10.1111/2041-210x.13945
- Population genomics of parallel hybrid zones in the mimetic butterflies, H. melpomene and H. erato.Genome Research24(8):1316‑1333. https://doi.org/10.1101/gr.169292.113
- Museum specimens: An overlooked and valuable material for conservation genetics.Ecological Research36(1):13‑23. https://doi.org/10.1111/1440-1703.12181
- Green Carbon, Black Trade: Illegal Logging, Tax Fraud and Laundering in the Worlds Tropical Forests. A Rapid Response Assessment.United Nations Environment Programme, GRIDArendal[ISBN9788277011028]
- Large scale genome skimming from herbarium material for accurate plant identification and phylogenomics.Plant Methods16:1. https://doi.org/10.1186/s13007-019-0534-5
- A high quality, high molecular weight DNA extraction method for PacBio HiFi genome sequencing of recalcitrant plants.Plant Methods19(1). https://doi.org/10.1186/s13007-023-01009-x
- Ancient DNA analysis.Nature Reviews Methods Primers1(1):1‑26. https://doi.org/10.1038/s43586-020-00011-0
- Not a limitless resource: ethics and guidelines for destructive sampling of archaeofaunal remains.Royal Society Open Science6(10):191059. https://doi.org/10.1098/rsos.191059
- Advantages of an easy-to-use DNA extraction method for minimal-destructive analysis of collection specimens.PLOS One15(7):e0235222. https://doi.org/10.1371/journal.pone.0235222
- Double digest RADseq: an inexpensive method for De Novo SNP discovery and genotyping in model and non-model species.PLOS One7(5):e37135. https://doi.org/10.1371/journal.pone.0037135
- Present-day DNA contamination in ancient DNA datasets.Bioessays42(9):2000081. https://doi.org/10.1002/bies.202000081
- nlme: Linear and nonlinear mixed effects Models. R package version 3.1-160. .
- Boots on the ground in Africa's ancient DNA ‘revolution’: archaeological perspectives on ethics and best practices.Antiquity92(363):803‑815. https://doi.org/10.15184/aqy.2018.70
- DNA barcodes from century-old type specimens using next-generation sequencing.Molecular Ecology Resources16(2):487‑497. https://doi.org/10.1111/1755-0998.12474
- Demystifying the RAD fad.Molecular Ecology23(24):5937‑5942. https://doi.org/10.1111/mec.12965
- BOLD: The Barcode of Life Data System (http://www.barcodinglife.org).Molecular Ecology Notes7(3):355‑364. https://doi.org/10.1111/j.1471-8286.2007.01678.x
- A DNA-based registry for all animal species: the barcode index number (BIN) system.PLOS One8(7):e66213. https://doi.org/10.1371/journal.pone.0066213
- Mining museums for historical DNA: advances and challenges in museomics.Trends in Ecology & Evolution36(11):1049‑1060. https://doi.org/10.1016/j.tree.2021.07.009
- Non-destructive extraction of DNA from preserved tissues in medical collections.BioTechniques72(2):60‑64. https://doi.org/10.2144/btn-2021-0014
- Authentication and assessment of contamination in ancient DNA.Methods in Molecular Biology1963:163‑194. https://doi.org/10.1007/978-1-4939-9176-1_17
- The importance of mycological and plant herbaria in tracking plant killers.Frontiers in Ecology and Evolution7:521. https://doi.org/10.3389/fevo.2019.00521
- On the causes, consequences, and avoidance of PCR duplicates: towards a theory of library complexity.Molecular Ecology Resources23:1299‑1318. https://doi.org/10.1111/1755-0998.13800
- Nondestructive DNA extraction method for mitochondrial DNA analyses of museum specimens.BioTechniques36(5):814‑821. https://doi.org/10.2144/04365ST05
- Sequence capture from historical museum specimens: maximizing value for population and phylogenomic studies.Frontiers in Ecology and Evolution10:931644. https://doi.org/10.3389/fevo.2022.931644
- Recovering the genomes hidden in museum wet collections.Molecular Ecology Resources22(6):2127‑2129. https://doi.org/10.1111/1755-0998.13631
- Characterizing restriction enzyme-associated loci in historic ragweed (Ambrosia artemisiifolia) voucher specimens using custom-designed RNA probes.Molecular Ecology Resources17(2):209‑220. https://doi.org/10.1111/1755-0998.12610
- Enhancing DNA barcode reference libraries by harvesting terrestrial arthropods at the National Museum of Natural History.Biodiversity Data Journal11:e100904. https://doi.org/10.3897/BDJ.11.e100904
- How to open the treasure chest? Optimising DNA extraction from herbarium specimens.PLOS One7(8):e43808. https://doi.org/10.1371/journal.pone.0043808
- Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA.PLOS One7(3):e34131. https://doi.org/10.1371/journal.pone.0034131
- Hy RAD ‐X, a versatile method combining exome capture and RAD sequencing to extract genomic information from ancient DNA.Methods in Ecology and Evolution8(10):1374‑1388. https://doi.org/10.1111/2041-210x.12785
- Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX.Nature Protocols9(5):1056‑1082. https://doi.org/10.1038/nprot.2014.063
- AdapterRemoval v2: rapid adapter trimming, identification, and read merging.BMC Research Notes9:88. https://doi.org/10.1186/s13104-016-1900-2
- Amplification of TruSeq ancient DNA libraries with AccuPrime Pfx: consequences on nucleotide misincorporation and methylation patterns.STAR: Science & Technology of Archaeological Research1(1):1‑9. https://doi.org/10.1179/2054892315Y.0000000005
- A non-destructive DNA sampling technique for herbarium specimens.PLOS One12(8):e0183555. https://doi.org/10.1371/journal.pone.0183555
- CPGAVAS2, an integrated plastome sequence annotator and analyzer.Nucleic Acids Research47(W1):W65‑W73. https://doi.org/10.1093/nar/gkz345
- Entomological collections in the age of big data.Annual Review of Entomology63:513‑530. https://doi.org/10.1146/annurev-ento-031616-035536
- Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal.Proceedings of the National Academy of Sciences of the United States of America111(6):2229‑2234. https://doi.org/10.1073/pnas.1318934111
- Efficiency of ddRAD target enriched sequencing across spiny rock lobster species (Palinuridae: Jasus).Scientific Reports7:6781. https://doi.org/10.1038/s41598-017-06582-5
- Isolation of high quality and polysaccharide-free DNA from leaves of Dimorphandra mollis (Leguminosae), a tree from the Brazilian Cerrado.Genetic and Molecular Research11(1):756‑764. https://doi.org/10.4238/2012.March.22.6
- The Rhododendron genome and chromosomal organization provide insight into shared whole-genome duplications across the heath family (Ericaceae).Genome Biology and Evolution11(12):3353‑3371. https://doi.org/10.1093/gbe/evz245
- A comparative study of RNA yields from museum specimens, including an optimized protocol for extracting RNA from formalin-fixed specimens.Frontiers in Ecology and Evolution10:953131. https://doi.org/10.3389/fevo.2022.953131
- DNA damage in plant herbarium tissue.PLOS One6(12):e28448. https://doi.org/10.1371/journal.pone.0028448
- Successful application of ancient DNA extraction and library construction protocols to museum wet collection specimens.Molecular Ecology Resources21(7):2299‑2315. https://doi.org/10.1111/1755-0998.13433
- Museomics for reconstructing historical floristic exchanges: Divergence of stone oaks across Wallacea.PLOS One15(5):e0232936. https://doi.org/10.1371/journal.pone.0232936
- Hybridization capture using RAD probes (hyRAD), a new tool for performing genomic analyses on collection specimens.PLOS One11(3):e0151651. https://doi.org/10.1371/journal.pone.0151651
- Non-destructive DNA extraction from herbarium specimens: a method particularly suitable for plants with small and fragile leaves.Journal of Plant Research133(1):133‑141. https://doi.org/10.1007/s10265-019-01152-4
- The effects of soft tissue removal methods on porcine skeletal remains.New Florida Journal of Anthropology1(2):30‑46. https://doi.org/10.32473/nfja.v1i2.124117
- Sequence locally, think globally: The Darwin Tree of Life Project.Proceedings of the National Academy of Sciences of the United States of America119(4):e2115642118. https://doi.org/10.1073/pnas.2115642118
- Rapid assembly of taxonomically validated mitochondrial genomes from historical insect collections.The Biological Journal of the Linnean Society117(1):83‑95. https://doi.org/10.1111/bij.12552
- Utility of arsenic-treated bird skins for DNA extraction.BMC Research Notes4:197. https://doi.org/10.1186/1756-0500-4-197
- Museomics of a rare taxon: placing Whalleyanidae in the Lepidoptera Tree of Life.Systematic Entomology46(4):926‑937. https://doi.org/10.1111/syen.12503
- Evolution at two time frames: Polymorphisms from an ancient singular divergence event fuel contemporary parallel evolution.PLOS Genetics14(11):e1007796. https://doi.org/10.1371/journal.pgen.1007796
- Index hopping on the Illumina HiseqX platform and its consequences for ancient DNA studies.Molecular Ecology Resources20(5):1171‑1181. https://doi.org/10.1111/1755-0998.13009
- Million-year-old DNA sheds light on the genomic history of mammoths.Nature591(7849):265‑269. https://doi.org/10.1038/s41586-021-03224-9
- The survival of PCR-amplifiable DNA in cow leather.Journal of Archaeological Science34(5):823‑829. https://doi.org/10.1016/j.jas.2006.09.002
- Back to the future: museum specimens in population genetics.Trends in Ecology & Evolution22(12):634‑642. https://doi.org/10.1016/j.tree.2007.08.017
- The Extended Specimen: Emerging Frontiers in Collections-Based Ornithological Research.CRC PressURL: https://play.google.com/store/books/details?id=sjsPEAAAQBAJ [ISBN9781498729161]
- Temporal patterns of damage and decay kinetics of DNA retrieved from plant herbarium specimens.Royal Society Open Science3(6):160239. https://doi.org/10.1098/rsos.160239
- Mapping the biosphere: exploring species to understand the origin, organization and sustainability of biodiversity.Systematics and Biodiversity10(1):1‑20. https://doi.org/10.1080/14772000.2012.665095
- Ancient DNA.Proceedings of the Royal Society B272(1558):3‑16. https://doi.org/10.1098/rspb.2004.2813
- Ancient biomolecules from deep ice cores reveal a forested southern Greenland.Science317(5834):111‑114. https://doi.org/10.1126/science.1141758
- Fast stable restricted maximum likelihood and marginal likelihood estimation of semiparametric generalized linear models.Journal of the Royal Statistical Society Series B73(1):3‑36. https://doi.org/10.1111/j.1467-9868.2010.00749.x
- Museums are biobanks: unlocking the genetic potential of the three billion specimens in the world's biological collections.Current Opinion in Insect Science18:83‑88. https://doi.org/10.1016/j.cois.2016.09.009
- Longer is not always better: optimizing barcode length for large-scale species discovery and identification.Systematic Biology69(5):999‑1015. https://doi.org/10.1093/sysbio/syaa014
- The rise and fall of the Phytophthora infestans lineage that triggered the Irish potato famine.Elife2:e00731. https://doi.org/10.7554/eLife.00731
- Genome skimming herbarium specimens for DNA barcoding and phylogenomics.Plant Methods14:43. https://doi.org/10.1186/s13007-018-0300-0
- PEAR: a fast and accurate Illumina Paired-End reAd mergeR.Bioinformatics30(5):614‑620. https://doi.org/10.1093/bioinformatics/btt593
- The draft genome assembly of Rhododendron delavayi var. delavayi.GigaScience6(10):1‑11. https://doi.org/10.1093/gigascience/gix076
Supplementary materials
List of equipment, its purpose and exemplar manufacturer, model and cost (as of 2022) for establishing an hDNA facility.
Spreadsheet containing sample ID, accession details, collecting data, preservation method, laboratory protocols used and DNA quantity recovery and sequence quality statistics.
Accession details and sample type of the specimens analysed in Case study 3.
Accession details, links to living and herbarium specimens databases, DNA concentration and library preparation methods.
Read statistics (raw reads, reads mapping to target loci, read clonality, coverage).
Sample information, DNA extracts evaluation and DNA reads description.