|
Biodiversity Data Journal :
Research Article
|
|
Corresponding author: Dongfang Xu (dongfangxu9@email.arizona.edu), Hong Cui (hongcui@email.arizona.edu)
Academic editor: Quentin Groom
Received: 14 May 2018 | Accepted: 27 Jul 2018 | Published: 10 Aug 2018
© 2018 Dongfang Xu, Steven Chong, Thomas Rodenhausen, Hong Cui
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation:
Xu D, Chong S, Rodenhausen T, Cui H (2018) Resolving “orphaned” non-specific structures using machine learning and natural language processing methods. Biodiversity Data Journal 6: e26659. https://doi.org/10.3897/BDJ.6.e26659
|
|
Abstract
Scholarly publications of biodiversity literature contain a vast amount of information in human readable format. The detailed morphological descriptions in these publications contain rich information that can be extracted to facilitate analysis and computational biology research. However, the idiosyncrasies of morphological descriptions still pose a number of challenges to machines. In this work, we demonstrate the use of two different approaches to resolve meronym (i.e. part-of) relations between anatomical parts and their anchor organs, including a syntactic rule-based approach and a SVM-based (support vector machine) method. Both methods made use of domain ontologies. We compared the two approaches with two other baseline methods and the evaluation results show the syntactic methods (92.1% F1 score) outperformed the SVM methods (80.7% F1 score) and the part-of ontologies were valuable knowledge sources for the task. It is notable that the mistakes made by the two approaches rarely overlapped. Additional tests will be conducted on the development version of the Explorer of Taxon Concepts toolkit before we make the functionality publicly available. Meanwhile, we will further investigate and leverage the complementary nature of the two approaches to further drive down the error rate, as in practical application, even a 1% error rate could lead to hundreds of errors.
Keywords
Information Extraction, Machine Learning, Anaphora Resolution, Ontology Application, Biodiversity Literature, Morphological Descriptions, Performance Evaluation
Background
Using the large volumes of information contained in biodiversity literature, we aim to provide scientists with rich computable data so they can build a more complete tree of life, predict the causal genes for wider ranges of diseases/conditions, derive better models of the decline of species populations and improve climate change predictions. Biodiversity literature contains various descriptive information on extinct and extant taxa, including habitat, distribution, phenology, ecology, physiology and morphology. In this work, we are primarily concerned with making the information contained in morphological descriptions more accessible to machines. One way to accomplish this is to extract character information from descriptions into a structured format, such as taxon-by-character matrices. This work involves linking a structure/organ (e.g. leaf or stem) with its properties (e.g. colour, shape, orientation etc., often called “characters” in systematics biology), as described in the literature (
In computational linguistics, this problem is treated as a subclass of the bridging reference problem, whose resolution requires not only finding the anchor in the text to which the bridging reference is related, but also identifying the relationship between them (
An illustrative example of our task is given below:
Leaflets articulated, inserted near the edges of the rhachis towards the adaxial side, lacking a differently coloured basal gland; stomata on lower surface only or on both surfaces; epidermal cells elongated parallel to long axes of leaflets.
In this example, the goal is to associate edges, adaxial side, lower surface, surfaces, cells and long axes with their anchors (parents) rhachis, leaflets, leaflets, leaflets, epidermal and leaflets, respectively.
This paper reports the evaluation of two methods we developed to resolve NSS in taxonomic descriptions. We start by giving a formal task definition and then, in the Data Resources section, we describe the corpus, datasets and ontologies used to develop and evaluate the methods. After introducing the two methods, we present evaluation results. We conclude the paper with future directions for research.
Task definition
Morphological descriptions of the biodiversity domain pose a number of challenges to existing natural language processing (NLP) algorithms.
Although natural language processing tools and algorithms have been successfully implemented in various tasks, bridging references are considered one of the most challenging problems in discourse analysis since the knowledge about the events and natural objects required to identify each relation goes beyond the text itself (
Given a short piece of text (one or a few sentences) from a morphological description that contains some NSS terms, our task was to resolve the NSSs, i.e. identify the anchor (parent) structure for each NSS. More formally, given a set of NSS-terms A = {a1, a2, a3,…, an} and a text segment W = {w1, w2, w3, ..., wn} consisting of n tokens, for any given wi=aj and, amongst all semantically valid part-of relations (wi, wk), our task was to find the specific wk that would resolve wi as the intended independent entity described by the description.
Data resources
A total of 7562 sentences from 3876 morphological description files of 11 taxon groups were extracted from various taxonomic data sources we have collected in prior years, including Plazi.org and the Flora of North America (FNA). A list of 39 NSS terms pertinent to these descriptions was created by domain experts. Table
|
NSS term |
count |
NSS term |
count |
NSS term |
count |
NSS term |
count |
|
apex |
511 |
centre |
52 |
groove |
350 |
protuberance |
8 |
|
appendix |
13 |
chamber |
14 |
layer |
25 |
remnant |
28 |
|
area |
476 |
component |
7 |
line |
163 |
section |
112 |
|
band |
114 |
concavity |
2 |
margin |
2419 |
side |
609 |
|
base |
684 |
content |
22 |
middle |
271 |
stratum |
2 |
|
belt |
2 |
crack |
8 |
notch |
80 |
surface |
959 |
|
body |
1171 |
edge |
258 |
part |
264 |
tip |
82 |
|
cavity |
148 |
element |
63 |
pore |
101 |
wall |
23 |
|
cell |
120 |
end |
166 |
portion |
240 |
zone |
51 |
|
centre |
49 |
face |
727 |
projection |
37 |
|
|
An initial dataset consisting of 169 sentences were randomly selected from taxonomic descriptions of ants, mushrooms and plants with 389 NSS term occurrences. This was used as the dataset to develop the two methods reported in this paper. To expand the taxon and NSS terms coverage, another 167 sentences were sampled in a stratified random manner from the corpus to form the test dataset; sentences were randomly selected from all 11 taxon groups and each sentence contained at least one NSS term. 366 NSS term occurrences were in the test dataset. Sentences containing target anchors were added if they were not sampled in the first round. After the data collection procedure, sentences that contained both NSS term(s) and target anchor(s) were treated as statements. The composition of the training and test datasets is shown in Table
|
Taxon |
Total Number of Sentences |
Development Dataset (# of statements) |
Test Dataset (# of statements) |
|
Ants |
866 |
21 |
20 |
|
Bees |
2189 |
18 |
21 |
|
Birds |
516 |
13 |
10 |
|
Diatoms |
503 |
13 |
18 |
|
Ferns |
630 |
15 |
7 |
|
FNA v-5 (pink, leadwort and buckwheat families) |
686 |
13 |
9 |
|
Gymnodinia |
206 |
10 |
13 |
|
Mushrooms |
815 |
26 |
35 |
|
Nematodes |
604 |
15 |
18 |
|
Sponges |
92 |
13 |
5 |
|
Weevils |
455 |
12 |
11 |
|
Total |
7562 |
169 |
167 |
Generation of the annotated evaluation datasets
The evaluation datasets (development and test) needed to be annotated with NSSs and their anchor organs. This was done semi-automatically. The fine-grained morphological character extraction system ETC Text Capture tool (
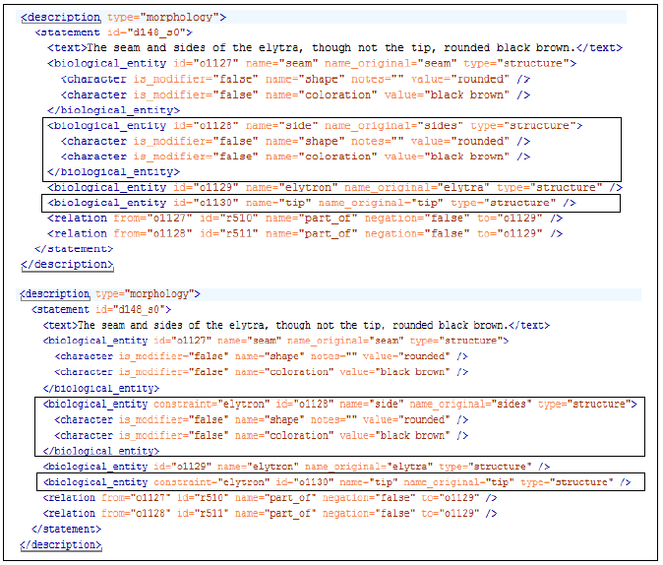
For performance evaluation, NSS-terms in both datasets were manually resolved by adding anchor organs to the NSS-terms. Fig.
The NSS Ontologies
Anatomical and phenotype character ontologies are being created by bioinformatics communities to generate computable data and support integration of data silos (
In this experiment, the tool was used by the same author who subsequently annotated the answer keys to create ontologies holding part-of relationships amongst various biological structures for the development and test data, respectively. Taking the statement in Fig. 1 as an example, we used the tool to first add the structures elytron, side, tip, elytron side and elytron tip into the ontologies and then linked the terms with part-of and subclass relationships (e.g. elytron side is a subclass of side and a part-of elytron). A two-month gap separated each of the ontology building events and gold standard annotation to control for any potential confounding between the ontologies and the gold standard. A total of 488/540 unique structures (including organ and NSS-terms) and 392/351 part-of relationships were created in the development/test versions of the NSS ontologies.
Methods
Syntactic rule method
This method took advantage of the syntactic parsing results embedded in the raw annotation of the input and utilised a set of simple rules to resolve the NSSs (i.e. finding the anchor organ for a NSS). Four rules were used: (1) biological entity terms within three-sentence boundaries of a NSS were considered candidate anchor organs (i.e. window size = 3); (2) part-of relationships generated by the ETC Text Capture tool through the of-phrases containing a NSS term, such as “base of the leaves”; (3) possession words around a NSS-term, such as “with”, “contain” and “have”; (4) the NSS ontologies.
Method 1 shows the pseudocode to obtain the mereological bridging references for NSS terms in each statement. First, it defined a queue to store all candidate anchor terms (i.e. biological entities, including all structure terms extracted by ETC) marked in one statement. It then sorted the NSS terms based on the order of their appearance in the queue and stored them into a list. For each NSS term in the list, it initialised a stack and pushed the current NSS term to the stack_NSS-to-be-resolved. The algorithm then used each rule in the list of rules [(2), (3), (1)] to find the candidate anchor term that satisfied one of the above rules (see #9 in pseudocode) and appeared in the ontology (#12). The list of rules [(2), (3), (1)] was ordered by their confidences (i.e. the order of confidences in rules for our task were: of-phrase > possession words > window size). Specifically, the algorithm first found any candidate anchor term in queue_terms that satistified both the of-phrase rule (2) and the ontology rule (4). If either rule was not satisfied, it continued checking rules (3) and (4) and then rules (1) and (4). The NSS-term was resolved if the above procedures returned any candidate anchor term, otherwise a null value was returned.
We also implemented a recursive approach to resolve bridging reference cases where multiple NSS terms were involved, such as “abdomen has a thin edge at its upper margin”; here, the anchor term of “edge” is “upper margin” which itself is also a NSS. To handle such situations, before the algorithm returns the candidate anchor term, it checks whether the candidate term is one of the NSS terms. If the answer is true, the algorithm then saves the relationship of the current NSS term and the candidate term (the new NSS term) and recursively finds the anchor term for the new NSS term until the new candidate anchor term is determined to be not a NSS. For the above example, the algorithm found the anchor term of “edge” to be "upper margin", which is also a NSS term. The algorithm then pushed the "upper margin" into the stack_NSS-to-be-resolved and resolved the new NSS term "upper margin". After finding the anchor term of "upper margin" to be "abdomen", the algorithm updated the anchor term of "edge" to be "abdomen upper margin".
#1 Initialize NSS Ontology, queue_terms, list_NSS-terms
#2 for entity in sentence do //store all biological entities: #2-#4
#3 insert entity into queue_terms
#4 end for
#5 list_NSS-terms = get_NSS(queue_terms) //obtain all NSS mentions
#6 for each NSS-term in list_NSS-terms do //resolve NSS term: #6-#28
#7 initialise stack_NSS-to-be-resolved
#8 push NSS-term to stack_NSS-to-be-resolved
#9 for each rule in rules [(2), (3), (1)] do
#10 for term in queue_terms do
#11 if rule(term, NSS-term) then //rules for terms: #11-#25
#12 if (term, NSS-term) in Ontology then
#13 if term is not in NSS-terms then//recursion: #13-23
#14 while stack_NSS-to-be-resolved is not empty do
#15 NSS-term ← pop(stack_NSS-to-be-resolved)
#16 anchor (NSS-term) ← term
#17 in list_NSS-terms delete NSS-term
#18 term ← term + NSS-term
#19 else:
#20 push term to stack_NSS-to-be-resolved
#21 NSS-term ← term
#22 goto #9
#23 end if
#24 end if
#25 end if
#26 end for
#27 end for
#28 end for
Method 1 Mereological Bridging Reference using Syntactic Rules and the NSS Ontologies.
This method and one variation of it were eventually evaluated. The variation resolves a NSS using a NSS ontology and the constraint of window size=3 (rules 1 and 4) and ignores the part-of and possession clues (rules 2 and 3). The source code for the Syntactic Method can be found at https://github.com/biosemantics/charaparser/tree/master/enhance.
Support vector machine
Support vector machine (SVM) functioning as a classifier remains an effective, low cost and robust method for many NLP tasks, especially for problems with a small number of training instances (
- Distance and position features: the indicator of whether the anchor term was the subject of a sentence/clause; the indicator of whether the anchor term was the closest term to the NSS term; absolute distance between the two structure terms in a statement; relative distance between the two structure terms in a statement (i.e. the absolute distance divided by the number of tokens in a statement); the absolute number of structure term(s) between the two structure terms; the relative number of structure term(s) between the two structure terms (i.e. the absolute number divided by the number of structures in a statement).
- Bag-of-word features: the indicators of whether the connectors between the term pairs were in the set of “in, on, at, of, has, have, with, contains, without”; tokens themselves before and after both structure terms in a certain context window, excluding the connectors listed above.
- Semantic features: the indicator of whether the term pairs appeared in the NSS ontology; the indicator of whether the anchor term was a NSS.
We used the LIBSVM implementation (
\(anchor\_term(NSS \,term)= argmax_{x ∈X} \, Pro(x,NSS \,term)\)
where \(X\) is the set of biological entity terms within one statement, excluding the NSS term which is to be resolved and \(Pro(x,NSS \,term)\) is a probability function which calculates the probability of \(x\) being the anchor term of the \(NSS \,term\). We used 5-fold cross validations to adjust the following parameters for the best performance: the word frequency threshold was set to 9 and the context window to 4 for the bag-of-word features; class-weight was set as 7 to handle the unbalanced classes; we experimented with multiple kernels and selected the polynomial kernel function with the degree set to 3; other parameters were set to their default.
This method and one variation of it were evaluated. The variation used features from groups 1 and 2, leaving out the semantic features. The source code of the SVM method can be found at https://github.com/biosemantics/SVM-for-Nonspecific-Structure.
Two baselines: subject entity and closest entity
Two additional baseline algorithms were also implemented and evaluated. For each NSS term, the first baseline algorithm (Baseline 1) always chose the subject entity term (i.e. the first entity appearing in a statement) as its anchor term, while the second baseline algorithm (Baseline 2) always identified the closest entity term around the NSS term as its anchor term. Taking the example from the Background section, NSS term edges would be linked to leaflets using the Baseline 1 algorithm or linked to rhachis using the Baseline 2 algorithm.
Evaluation metrics
The four methods and two variations were evaluated using precision, recall and f1 metrics, which are routinely used in information retrieval and information extraction tasks. Precision (P), recall (R) and f1 (F1) are formally defined below:
\(P(S, H) = \frac {|S ∩ H|} {|S| }\)
\(R(S, H) = \frac {|S ∩ H|} {|H|}\)
\( F1(S, H) = \frac {2 * P(S,H) *R(S,H)} { P(S,H) + R(S,H) }\)
where S is the set of anchor terms identified by the system and H is the set of anchor terms annotated in the gold standard.
Results and discussion
We compared the different methods described in the Methods section. Table
Performances of the Baseline, Syntactic and SVM Methods Using the Development and Test Datasets.
|
Methods |
F1 (Development) |
P (Test) |
R (Test) |
F1 (Test) |
|
Baseline 1 (subject entity) |
63.9% |
42.3% |
42.3% |
42.3% |
|
Baseline 2 (closest entity) |
30.3% |
33.2% |
33.2% |
33.2% |
|
Syntactic (ontology only) |
91.1% |
92.2% |
90.5% |
91.4% |
|
Syntactic (all rules) |
93.7% |
93.0% |
91.3% |
92.1% |
|
SVM (feature groups 1 and 2) |
76.1% |
60.9% |
60.9% |
60.9% |
|
SVM (all features) |
89.6% |
80.7% |
80.7% |
80.7% |
Second, we found the syntactic approach outperformed the SVM method by a large margin. For example, the best syntactic approach achieved an F1 score of 92.1%, while the best SVM model achieved an F1 of only 80.7%. This may be due to the small training size provided to the SVM. From a practical point of view, it was not realistic to request that domain scientists annotate large sets of training examples. In contrast, they are more likely to construct an ontology that can be reused to process future descriptions and solve other related problems, such as extracting phenotypes.
Third, we found ontologies were reliable knowledge sources in resolving orphaned parts in morphological descriptions. Using an ontology alone, the syntactic rule-based approach achieved an F1 score of 91.4%. Using an ontology also improved SVM performance by 20 percentage points for the F1 score. The syntactic rules (of-phrases and possession words) seem to be useful for the rule-based method, but using these rules with an ontology only improved the F1 score by 0.7% and, when considering the 366 test examples, their effects on reference resolution were unclear since the results may have been confounded by usage of an ontology. We are planning to evaluate a second variation of the rule-based method using only the rules and not an ontology to further examine the effects of these rules.
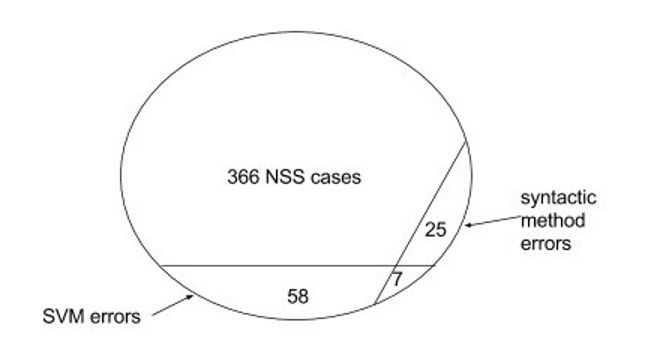
Our error analysis showed that the syntactic method and the SVM method were complementary to each other, as the mistakes made by the two methods were largely disjoint. Fig.
Conclusions and future work
With the results from the dataset covering a wide range of descriptions, we can tentatively conclude that the syntactic rule method is what the ETC toolkit will adopt, given that ETC toolkit already provides a user-friendly ontology building tool for domain scientists and students to create ontologies. However, in the immediate future, it is worthwhile for additional testing on the development version of ETC toolkit before we make the functionality publicly available and to further investigate and leverage the complementary nature of the syntactic rule method and the SVM method to minimise the error rate, as in practical application NSS, terms are commonly used in morphological descriptions and even a 1% error rate could lead to numerous errors.
Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. NSF DBI-1147266. The authors also thank anonymous reviewers for their constructive suggestions that helped to improve this paper.
References
- Bunescu R (2003) Associative anaphora resolution: A web-based approach. Proceedings of the EACL Workshop on the Computational Treatment of Anaphora.Budapest,47-52pp.
- LIBSVM: a library for support vector machines.ACM Transactions on Intelligent Systems and Technology (TIST)2(3):1‑39. https://doi.org/10.1145/1961189.1961199
- Gene name ambiguity of eukaryotic nomenclatures.Bioinformatics21(2):248‑256. https://doi.org/10.1093/bioinformatics/bth496
- Bottom-up ontology building for domains without existing standards.iConference 2017.iConference 2017 Proceedings,861-864pp. https://doi.org/10.9776/17356
- Bridging.Proceedings of the 1975 workshop on Theoretical issues in natural language processing - TINLAP '75https://doi.org/10.3115/980190.980237
- Semantic annotation of morphological descriptions: an overall strategy.BMC Bioinformatics11(1):278. https://doi.org/10.1186/1471-2105-11-278
- Introducing Explorer of Taxon Concepts with a case study on spider measurement matrix building.BMC Bioinformatics17(1):471. https://doi.org/10.1186/s12859-016-1352-7
- Toward Synthesizing Our Knowledge of Morphology: Using Ontologies and Machine Reasoning to Extract Presence/Absence Evolutionary Phenotypes across Studies.Systematic Biology64(6):936‑952. https://doi.org/10.1093/sysbio/syv031
- Extraction of phenotypic traits from taxonomic descriptions for the tree of life using natural language processing.Applications in Plant Sciences6(3):e1035. https://doi.org/10.1002/aps3.1035
- Indirect anaphora resolution as semantic path search.Proceedings of the 3rd International Conference on Knowledge Capture, ACM,153-160pp. https://doi.org/10.1145/1088622.1088650
- Definiteness and the processing of noun phrases in natural discourse.Journal of Semantics7(4):395‑433. https://doi.org/10.1093/jos/7.4.395
- The representation and use of focus in dialogue understanding.SRI International Menlo Park United States. Doctoral dissertation, University of California, Berkeley,170pp.
- Definiteness and indefiniteness.Croom Helm,London,316pp.
- The flora phenotype ontology (FLOPO): tool for integrating morphological traits and phenotypes of vascular plants.Journal of Biomedical Semantics7(1):65. https://doi.org/10.1186/s13326-016-0107-8
- OTO: Ontology Term Organizer.BMC Bioinformatics16(1). https://doi.org/10.1186/s12859-015-0488-1
- The Human Phenotype Ontology in 2017.Nucleic Acids Research45:D865‑D876. https://doi.org/10.1093/nar/gkw1039
- Document-level sentiment classification: An empirical comparison between SVM and ANN.Expert Systems with Applications40(2):621‑633. https://doi.org/10.1016/j.eswa.2012.07.059
- Corpus-based investigation of definite description use.Computational Linguistics24(2):183‑216.
- Learning to resolve bridging references.Proceedings of the 42nd Annual Meeting on Association for Computational Linguistics, Association for Computational Linguistics,143pp.
- Sindner CL (1979) Towards a Computational Theory of Definite Anaphora Comprehension in English Discourse.Massachusetts Institute of TechnologyURL: http://hdl.handle.net/1721.1/16010
- Applications of natural language processing in biodiversity science.Advances in Bioinformatics2012:1‑17. https://doi.org/10.1155/2012/391574
- Proceedings of COLING 2014, the 25th International Conference on Computational Linguistics: Technical Papers.COLING 2014, the 25th International Conference on Computational Linguistics,Dublin, Ireland,August 23-29 2014.2335–2344pp.