|
Biodiversity Data Journal :
Forum Paper
|
|
Corresponding author: Hong Cui (hongcui@email.arizona.edu)
Academic editor: Sarah Faulwetter
Received: 07 Sep 2018 | Accepted: 23 Oct 2018 | Published: 07 Nov 2018
This is an open access article distributed under the terms of the CC0 Public Domain Dedication.
Citation:
Cui H, Macklin J, Sachs J, Reznicek A, Starr J, Ford B, Penev L, Chen H (2018) Incentivising use of structured language in biological descriptions: Author-driven phenotype data and ontology production. Biodiversity Data Journal 6: e29616. https://doi.org/10.3897/BDJ.6.e29616
|
|
Abstract
Phenotypes are used for a multitude of purposes such as defining species, reconstructing phylogenies, diagnosing diseases or improving crop and animal productivity, but most of this phenotypic data is published in free-text narratives that are not computable. This means that the complex relationship between the genome, the environment and phenotypes is largely inaccessible to analysis and important questions related to the evolution of organisms, their diseases or their response to climate change cannot be fully addressed. It takes great effort to manually convert free-text narratives to a computable format before they can be used in large-scale analyses. We argue that this manual curation approach is not a sustainable solution to produce computable phenotypic data for three reasons: 1) it does not scale to all of biodiversity; 2) it does not stop the publication of free-text phenotypes that will continue to need manual curation in the future and, most importantly, 3) It does not solve the problem of inter-curator variation (curators interpret/convert a phenotype differently from each other). Our empirical studies have shown that inter-curator variation is as high as 40% even within a single project. With this level of variation, it is difficult to imagine that data integrated from multiple curation projects can be of high quality. The key causes of this variation have been identified as semantic vagueness in original phenotype descriptions and difficulties in using standardised vocabularies (ontologies). We argue that the authors describing phenotypes are the key to the solution. Given the right tools and appropriate attribution, the authors should be in charge of developing a project’s semantics and ontology. This will speed up ontology development and improve the semantic clarity of phenotype descriptions from the moment of publication. A proof of concept project on this idea was funded by NSF ABI in July 2017. We seek readers input or critique of the proposed approaches to help achieve community-based computable phenotype data production in the near future. Results from this project will be accessible through https://biosemantics.github.io/author-driven-production.
Keywords
Controlled Vocabulary, Computable Phenotype Data, Data Quality, Phenotype Ontologies
Introduction
Phenotypes are paramount for describing species, studying function and understanding organismal evolution. Recent advancements in computation technology have enabled large-scale, data-driven research, but its full potential has not been realised due to lack of data. High impact research, such as studying trait evolution and its relationship to phylogeny and the environment (e.g.
Textual phenotype descriptions that hold valuable information are continuously being published, yet they are not amenable to computation. When added to the massive amount of phenotype data sitting in older publications, these free-text character descriptions represent a major, under-utilised resource for integrating phenotypic data into modern, large-scale biological research projects that typically involve genomic, climatic and habitat data. These descriptive data are often variable in expression and terminology. Different descriptions of the same character may appear to describe two different traits or two different characters might be interpreted as one. Transforming various natural language expressions into computable data requires a process, called ontologising, where the semantics (meaning) of varied expressions are mapped to terms in an ontology and therefore made explicit (
Currently, making free-text phenotype information computable requires highly trained post-doctoral researchers manually ontologising the descriptions, facilitated by some software applications. However, the manual curation of legacy descriptions is not a sustainable solution for phenotype data production because it does not stop the continued publication of free-text phenotype descriptions that need semantic curation before use. If we assume that each of the estimated 750,000 biomedical papers published in English in 2014 (
Manual curation also does not address the fundamental causes of large (~ 40%) variations in the phenotype data manually curated by different workers (e.g.
As long as phenotype descriptions continue to be produced as free text, computable phenotype data will remain a major bottleneck holding back large-scale biological research. Given the varied usages of phenotype terms/expressions by different authors and given the fact that the meanings of a term evolve over time, it is evident the semantics of phenotypic characters (categorical or continuous characters) can be most accurately captured at the time of writing by their authors. Any downstream process risks information loss or even misinformation.
Author-Driven Phenotype Data and Ontology Production
We have been awarded funding to investigate a new paradigm of phenotype data production centred on description authors and supported by intuitive software tools to allow them to compose semantically clear descriptions while contributing their vocabularies/expressions to a shared ontology for their taxon groups. It brings authors to the forefront of ontology construction, promotes clear expressions and exposure of all valid meanings of technical terms and encourages open collaboration and consensus building amongst scientists. While the proposed approach presents a major conceptual change in phenotype data authoring, the change can be introduced via software environments with which authors are already familiar, for example, Google Docs and Wikis. We will approach the project from the perspectives of social and software engineering, examining human social and collaborative behaviour (e.g. attribution and motivation) and software usability to identify factors that encourage or discourage users from adopting the approach. Although we will start with a test case using the plant genus Carex L. (“sedges”, family Cyperaceae), the project has the potential to change how biodiversity is described in general and dramatically ease the production of computable phenotype data at a large scale.
Using the ongoing Carex revisionary work as the evaluation case for this project is an excellent choice because: 1). Carex is one of the largest genera in flowering plants, with close to 2000 species containing considerable variation. 2). A network of Carex experts already work closely to prepare the revisions. 3). Carex is treated in Flora of North America and Flora of China, from which we have previously extracted over 1200 Carex morphological terms and will be used to build the initial Carex Phenotype Ontology (CPO) for scientists to improve and 4). Scientists on this project will use the large amount of characters produced from this approach to expand their past research to a scale not possible before (
We also note that the larger academic and scientific research environment support the premises of the proposed approach. The importance of computable phenotype data is widely recognised and data silos are being actively dissolved. Ontologies and other data publications are valued and attribution methods are being actively examined to credit intellectual contributions to digital resource curation, such as the efforts by the International Society for Curation (http://biocuration.org), OBO Foundry (http://www.obofoundry.org) and THOR (http://project-thor.eu). Publishers like Pensoft are actively seeking and welcoming new methods to stop the continued publication of legacy descriptions. Having years of experience with using digital tools/devices, scientists are expert users of digital collaborative environments (e.g. Wikis, Google Docs). The time is right to investigate a long-term solution to phenotype data production.
Proposed System Design
Fig.
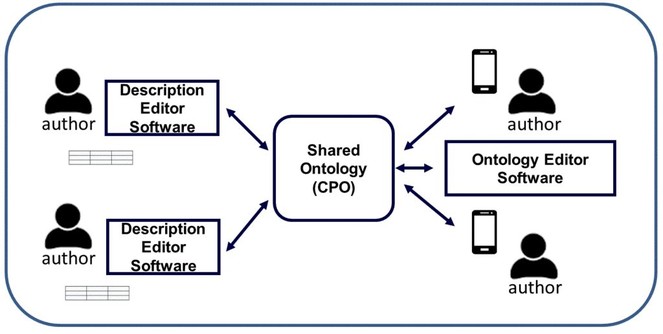
It is important to differentiate this approach from a standardisation approach where the authors are limited to using a set of “standardised” terms selected by others. The proposed approach does not limit author's choices, but it requires the authors to register the meaning (i.e. semantics) of the terms in their descriptions in an ontology and relate them to other existing terms to allow accurate interpretations in the future. For example, a standardisation approach might require Joe to use the term strong when he wishes to say stout. In contrast, our approach might show Joe that stout has two related but different meanings: increased size and strong (not fragile). This would allow Joe to choose the most precise term to use, increased size, strong or stout and, in turn, allow the reader, human or computer, to obtain the accurate meaning intended by the author. The key idea of the proposed approach is to make all valid meanings of a term clear and visible to a community of users and to encourage the user to filter and choose terms with the most accurate meaning for their purposes.
When the user adds a term to the ontology, the online open Ontology Editor is invoked, presenting different patterns to relate the terms in semantic ways (e.g. assert utricle in Carex ≡ perigynium in Carex, spike is_a inflorescence, spikelet ≡ secondary spike or small spike, stout ≡strong and increased size, weak ≡decreased magnitude or decreased strength). Ontology design patterns (e.g.
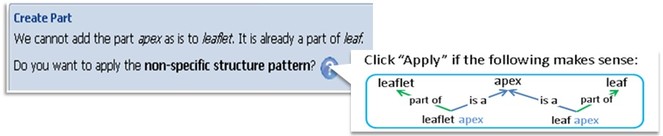
The system detects that the user is attempting to add a substructure (apex) to multiple parent structures (leaf and leaflet). This triggers the system to suggest the non-specific structure pattern to the user. When the user confirms, the system will insert four assertions (4 links in the graph) into the ontology automatically.
These patterns are expected to greatly improve the predictability of the ontology, reduce variation and lower the barrier to entry for biologists. The software will auto-detect situations, whenever possible, for which a pattern may be useful; once the user confirms, the system will carry out what needs to be done on the user’s behalf. Fig.
Small ontology building tasks such as conflicts amongst term definitions and relationships can be broadcast via a simple mobile app for registered authors to resolve at their leisure. Technical challenging cases can be resolved with help from trained ontology engineers, for example, the Planteome Project (http://planteome.org) or the OBO Foundry.
The rewards to authors who adopt this new workflow include: (1) Narrative descriptions in camera-ready form for publication. (2) A taxon-by-character matrix formulated ontology terms, ready for publication. These can be published in partner journals (e.g.Pensoft journals) in a customisable human readable form (e.g. sentences or matrices) and a variety of new ontologised formats such as EQs (Entity Quality) in the Phenoscape Knowledgebase (http://kb.phenoscape.org) or RDF graphs (Resource Description Framework, a format used widely on the Semantic Web). (3) Formal attributions and increased citations. On one hand, research has shown that studies that make their data available receive more citations than similar studies that do not (e.g.
Results from social and behavioural sciences research on computer mediated collaborative work, online community building and consensus making (e.g.
Expected Results
We hypothesise that, with careful design of the user interface that takes into account user-friendliness, efficiency, user motivation and other social and behavioural factors, this approach will increase phenotype data quality, ontology quality and computation efficiency.
-
Data quality: improve the semantic clarity of new phenotype descriptions to dramatically reduce the scope of the subsequent ontologisation effort,
-
Ontology quality: quickly improve the coverage of the phenotype ontology for a particular domain (e.g. a taxonomic group) and
-
Computation efficiency: obtain ontologised matrices and/or EQ statements with higher consistency and hence support a wide range of applications.
Assuming this proof of concept system is successful, this approach can be applied to any other science and engineering domains (e.g. biomedical, geology, astrophysics etc.). This being so, individual domain ontologies can be linked, based on shared concepts and terms, thus building powerful bridges for integration across domains, sciences and beyond.
Conclusion
Readers interested in learning more about our project and eventually evaluating our software prototypes can obtain further information from our github project page (https://biosemantics.github.io/author-driven-production) or contact authors. In summary, the goal of this project is to investigate the feasibility of transforming phenotype authors’ writing practice to produce computable phenotype data at the time of publication, with increased speed, scale, quality and consistency, while collectively curating phenotype ontologies, making them reflect a community consensus. Through thorough user experience research, we will also identify ways to reduce the entry barrier and promote user adoption of the new practice. When publishers adopt this new idea, we believe the ultimate goal of producing massive high-quality phenotype data for the entire scientific community can be achieved. We seek readers input or critique of the proposed approaches to help achieve community-based computable phenotype data production in the near future.
Grant title
ABI innovation: Authors in the Driver's Seat: Fast, Consistent, Computable Phenotype Data and Ontology Production
Hosting institution
University of Arizona
Author contributions
Cui, Macklin and Sachs contributed the initial idea of the project. All authors edited and reviewed the manuscript.
Conflicts of interest
The authors have declared that no competing interests exist.
References
- Berez-Kroeker A, Holton G, Kung S, Pulsifer P (2017) Developing standards for data citation and attribution for reproducible research in linguistics: project summary and next steps. Presentations from the Linguistic Society of America symposium and poster session on Data Citation and Attribution in Linguistics.Austin TX
- The Tenth Anniversary of Assigning DOI Names to Scientific Data and a Five Year History of DataCite.D-Lib Magazine21https://doi.org/10.1045/january2015-brase
- Proceedings of the ACM Conference on Computer Supported Cooperative Work.Association for Computing Machinery,New York,107-116pp.
- Implementation of data citations and persistent identifiers at the ORNL DAAC.Ecological Informatics33:10‑16. https://doi.org/10.1016/j.ecoinf.2016.03.003
- Proceedings of the 78th Annual Meeting of Association for Information Science and Technology.St. Louis, Missouri
- Introducing Explorer of Taxon Concepts with a case study on spider measurement matrix building.BMC Bioinformatics17:471. https://doi.org/10.1186/s12859-016-1352-7
- Moving the mountain: analysis of the effort required to transform comparative anatomy into computable anatomy.Database2015https://doi.org/10.1093/database/bav040
- Phenoscape: Identifying candidate genes for evolutionary phenotypes.Molecular Biology and Evolution33(1):13‑24. https://doi.org/10.1093/molbev/msv223
- Egaña M, Rector A, Stevens R, Antezana E (2008) Applying ontology design patterns in bio-ontologies. In: Gangemi A, Euzenat J (Eds) Knowledge Engineering: Practice and Patterns.Springer Berlin,Heidelberg,7-16pp.
- Reasoning over taxonomic change: Exploring alignments for the Perelleschus use case.PLOS ONE10(2):e0118247. https://doi.org/10.1371/journal.pone.0118247
- Proceedings of Computer-Supported Cooperative Work.Association for Computing Machinery,New York,85-93pp.
- Groupware and social dynamics: Eight challenges for developers.Communications of the ACM37(1):93‑104.
- Incorporating Data Citation in a Biomedical Repository: An Implementation Use Case.AMIA Joint Summits on Translational Science proceedings. AMIA Joint Summits on Translational Science2017:131‑138.
- Proceedings of the 7th International Symposium on Wikis and Open Collaboration.Association for Computing Machinery,New York,163-172pp.
- Snuggle: designing for efficient socialization and ideological critique.Proceedings of the 32nd Annual ACM Conference on Human Factors in Computing Systems - CHI '14,311-320pp. https://doi.org/10.1145/2556288.2557313
- OTO: Ontology Term Organizer.BMC Bioinformatics16:47. https://doi.org/10.1186/s12859-015-0488-1
- Consensus Building and Complex Adaptive Systems.Journal of the American Planning Association65(4):412‑423. https://doi.org/10.1080/01944369908976071
- Failure of large transformation projects from the viewpoint of complex adaptive systems: Management principles for dealing with project dynamics.Information Systems Frontiers17(1):15‑29. https://doi.org/10.1007/s10796-014-9511-8
- Proceedings of 27th International Conference on Human Factors in Computing Systems.Association for Computing Machinery,New York,1485-1494pp.
- Proceedings of GROUP: ACM Conference on Supporting Group Work.Association for Computing Machinery,New York,167-176pp.
- Proceedings of the ACM Conference on Human Factors in Computing Systems.Association for Computing Machinery,New York,1927-1936pp.
- Proceedings of the ACM Conference on Computer-Supported Cooperative Work.1007-1022pp.
- Phenotype ontologies: the bridge between genomics and evolution.Trends in Ecology & Evolution22(7):345‑350. https://doi.org/10.1016/j.tree.2007.03.013
- Annotation of phenotypes using ontologies: a Gold Standard for the training and evaluation of natural language processing systems.Database.
- An experiment-based methodology to understand the dynamics of group decision making.Socio-Economic Planning Sciences56:14‑26. https://doi.org/10.1016/j.seps.2016.06.001
- Editing beyond articles: diversity & dynamics of teamwork in open collaborations.Proceedings of the 17th ACM Conference on Computer Supported Cooperative Work & Social Computing - CSCW '14https://doi.org/10.1145/2531602.2531654
- Proceedings of the ACM Conference on Human Factors in Computing Systems.Association for Computing Machinery,New York,1917-1926pp.
- Data Citation Index.Journal of the Medical Library Association : JMLA104(1):88‑90. https://doi.org/10.3163/1536-5050.104.1.020
- Pender J (2016) Climatic niche estimation, trait evolution and species richness in North American Carex (Cyperaceae) (M.Sc. Thesis).University of Ottawa
- Data reuse and the open data citation advantage.PeerJ1(175). URL: https://doi.org/10.7717/peerj.175
- The reader-to-leader framework: Motivating technology mediated social participation.AIS Transactions on Human-Computer Interaction1(1):13‑32. https://doi.org/10.17705/1thci.00005
- Presutti V, Blomqvist E, Daga E, Gangemi A (2012) Pattern-Based Ontology Design. In: Suárez-Figueroa MC, Gómez-Pérez A, Motta E, Gangemi A (Eds) Ontology Engineering in a Networked World.Springer,New York,35-64pp.
- Analyzing data citation practices using the data citation index.Journal of the Association for Information Science and Technology67(12):2964‑2975. https://doi.org/10.1002/asi.23529
- Social barriers faced by newcomers placing their first contribution in open source software projects.Proceedings of the 18th ACM Conference on Computer Supported Cooperative Work & Social Computing - CSCW '15https://doi.org/10.1145/2675133.2675215
- The STM Report: An Overview of Scientific and Scholarly Journal Publishing.4th Edition.International Association of STM Publishers,The Hague,180pp.
- Proceedings of the 9th ACM Conference on Electronic Commerce.Association for Computing Machinery,New York,302-309pp.
- Proceedings of the 2012 ACM Conference on Human Factors in Computing Systems.Association for Computing Machinery,New York,2905-2914pp.
- Three keys to the radiation of angiosperms into freezing environments.Nature506(7486):89‑92. https://doi.org/10.1038/nature12872
- Proceedings of the ACM Conference on Computer Supported Cooperative Work.Association for Computing Machinery,New York,407-416pp.