|
Biodiversity Data Journal :
Research Article
|
|
Corresponding author: Anila Fariq (neelaahmad@gmail.com), Azra Yasmin (azrayasmin@fjwu.edu.pk)
Academic editor: Chloe Robinson
Received: 20 May 2021 | Accepted: 19 Jul 2021 | Published: 20 Oct 2021
© 2021 Anila Fariq, Azra Yasmin, John Blazier, Sammyia Jannat
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation:
Fariq A, Yasmin A, Blazier JC, Jannat S (2021) Identification of bacterial communities in extreme sites of Pakistan using high throughput barcoded amplicon sequencing. Biodiversity Data Journal 9: e68929. https://doi.org/10.3897/BDJ.9.e68929
|
|
Abstract
Microorganisms thrive nearly everywhere including extreme environments where few other forms of life can exist. Geochemistry of extreme sites plays a major role in shaping these microbial communities and microbes thriving in such harsh conditions are untapped sources of novel biomolecules. To understand the structure and composition of such microbial communities, culture-independent bacterial diversity was characterised for two extreme sites in Pakistan, Khewra salt range and Murtazaabad hot spring. Barcoded amplicon sequencing technique was used to study the microbial communities. Physicochemical analysis of these sites was also conducted to study the dynamics of microbial communities under stressed conditions. Metagenomic sequencing of salt range soil samples yielded of 40,433 16S rRNA sequences, while hot spring sediments produced 76,449 16S rRNA sequence reads. Proteobacteria were predominant in saline soil while Firmicutes were most abundant in hot spring sediment. The taxonomic analysis of saline samples revealed 914 operational taxonomic units (OTUs) while that of hot spring sequences were clustered into 726 distinct OTUs. OTUs from genus Alkalibacillus were most abundant in hot spring sediments, whereas Haloarcula were more prevalent in saline soil. Some unidentified sequences were also present at each taxonomic level. Multivariate analysis indicated that electrical conductivity and pH are the major environmental factors involved in modelling microbial communities. This study revealed a poly-extremophilic microbial community in the Murtazaabad hot spring and characterised the unexplored halophilic microbial diversity of saline soil of Pakistan.
Keywords
culture independent, barcoded amplicon sequencing, operational taxonomic units, poly-extremophilic
Introduction
Many prokaryotes reside in extreme environments in which some chemical or physical environmental parameters vary considerably from the normal habitats that support life. Such organisms are called extremophiles, flourishing in habitats which are hostile for other living organisms. Isolation and characterisation of extremophilic prokaryotes in recent years revealed their metabolic potential (
The development of molecular tools and the ability to isolate microbes in laboratory culture have revolutionised knowledge of bacterial diversity, which greatly exceeds that found in eukaryotes. There is currently great interest in mining the genetic resources of prokaryotic cells to be used in biotechnology and related areas (
Modern metagenomic studies, like 16S rRNA sequencing, accurately characterise microbial diversity. However, many types of locale are still un-sampled and extensive effort is required to determine the patterns of microbial ecology and evolution in such extreme environments. Nowadays, these communities have gained more attention in applied research both due to their biotechnological potential and to comprehend the evolution of biomolecules from their analogues found in other organisms (
A study reported the microbial diversity of three hot springs from Neuquén, Argentina, by using molecular based high-throughput amplicon sequencing technique. This study demonstrated metabolic profiling in the acidic and the circum-neutral samples as the former were dominated by chemolithotrophs, while the latter were dominated by chemoheterotrophs. The research also described that microbial communities were shaped by complex factors other than pH and temperature (
Pakistan is situated in the sub-continent along the junctions of the tectonic plates and is rich in geothermal resources. The present geological structure of Pakistan was formed by the major tectonic elements in the Cenozoic and Mesozoic era. The distribution of hot springs reflects the movements of tectonic plates. These hot springs are distributed in Chilas and Hunza along the plunge of main mantle and Karakoram. Temperature of these hot springs may reach 96°C (
The present study aims to investigate the composition of bacterial communities of extreme sites of Pakistan including Murtazabad hot spring and Khewra salt range by using Bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) (
Materials and methods
Sample collection
The saline soil sample was collected from Khewra salt range at the latitude and longitude of 32ºN, 73ºE, while the sediment sample was collected from the hot spring of Murtazaabad, located in Gilgit Baltistan at 35ºN and 76º E, respectively. Six soil samples from each site were collected at a depth of 0 to 5 cm by metallic tubes with a diameter of 5 cm, mixed evenly and placed in sterile zipper plastic storage bags.
Physicochemical analysis
To determine the electrical conductivity and pH of samples, aqueous solutions of samples were prepared in the ratio of 1:10 and electrical conductivity was measured by EC meter, while pH was measured by pH meter. For metal analysis, soil was digested by the acid digestion method (
Barcoded amplicon sequencing
Total soil DNA was extracted with QIAGEN PowerSoil kit by following standard protocols. Barcoded amplicon sequencing (bTEFAP®) was performed by MR DNA as described by
Data analysis
The QIIME data analysis package was used for 16S rRNA data analysis (
Results
Geochemistry of the saline sample showed neutral pH and very high electrical conductivity (19 mS/cm) reflecting the hypersaline nature of the sample. The hot spring sediment sample exhibited alkaline pH and conductivity of 2.17 mS/cm. Both samples contained variable concentrations of different heavy metals i.e. Pb, Ni, Cu, Zn, Cd and large concentrations of Ca, K and Na (Table
|
Parameters |
Saline soil |
Hot spring sediment |
|
Temperature (°C) |
28 |
50 |
|
pH |
7.28 |
7.8 |
|
Conductivty (mS/cm) |
19 |
2.17 |
|
Pb (mg/kg) |
182.5 |
12.015 |
|
Ni (mg/kg) |
113.5 |
20.7 |
|
Cu (mg/kg) |
108.83 |
32.56 |
|
Zn (mg/kg) |
72.8 |
21.1 |
|
Cd (mg/kg) |
28.35 |
2.76 |
|
Na(mg/kg) |
10816 |
5550 |
|
Ca (mg/kg) |
11475 |
600 |
|
K (mg/kg) |
6400 |
9150 |
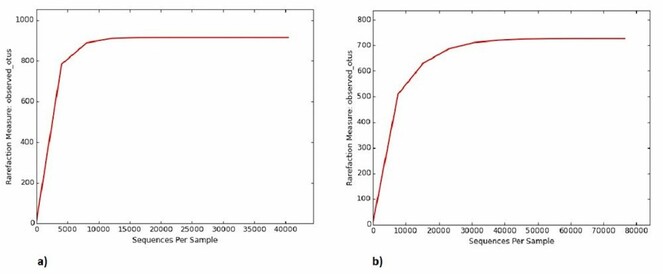
G+C content of the saline sample was about 56% and that of the sediment sample was 56.5%. About 40,433 high quality 16S rRNA sequences obtained from saline sample and 76,449 from sediment sample were clustered into 914 and 726 operational taxonomic units (OTUs), respectively after filtering low-confidence sequences with fewer than three counts. Rarefaction curves estimated the diversity captured in each sample (Fig.
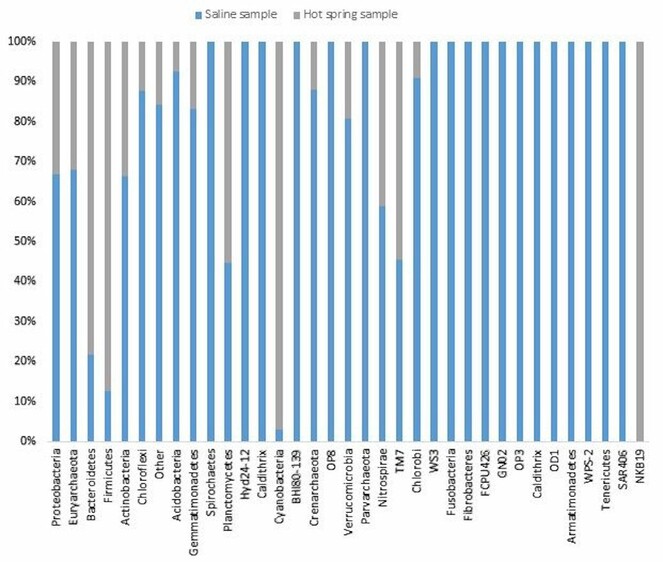
In the saline sample, a total of 35 distinct prokaryotic phyla were detected by metagenomic analysis, out of which Proteobacteria account for 46.20% of the total biodiversity. Amongst Proteobacteria, Gammaproteobacteria was the most prevalent class of bacteria. Deltaproteobacteria was comparatively less abundant. Alphaproteobacteria and Betaproteobacteria were poorly represented. Euryarchaeota was the second most abundant phylum and contributed to 25.38% of diversity. In archaea, Halobacteria was the most predominant class, as expected in an extreme halophilic community. Other relatively less abundant phyla found in hypersaline soil included Actinobactria (3.53%), Bacteroidetes (4.24%), Firmicutes (4.14%), Acidobacteria (2.24%), Chloroflexi (2.85%), Gemmatimonadetes (2.10%), Spirochetes (1.64%) and Cyanobacteria (0.41%). About 2.47% of the total phyla were not assigned to any category and classified as “other”. In the hot spring sample, 19 different phyla were identified. Firmicutes, Proteobacteria, Bacteroidetes, Cyanobacteria and Euryarchaeota constituted the major components with 29.14, 22.81, 15.29, 13.77 and 12.08%, respectively of total bacterial population. Other relatively less dominant phyla include Actinobacteria, Planctomycetes, Gemmatimonadetes, Chloroflexi, Acidobacteria, Crenarchaeota and Nitrospirae (Fig.
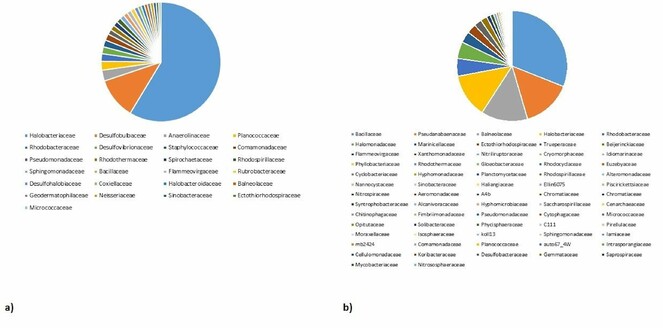
At the family level in the saline sample, Halobacteriaceae is the most predominant family displaying 25.19% of the total population. Other families identified in the metagenomic data (Fig.
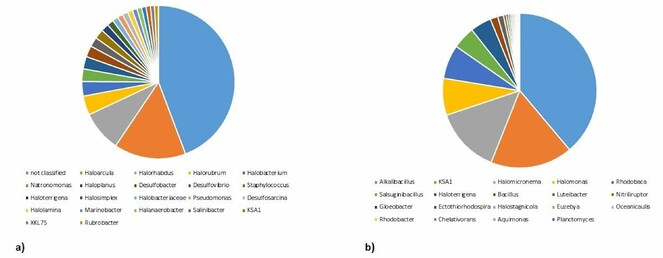
In the saline sample, at the genus level, Haloarcula, Halorhabdus, Halorubrum, Halobacterium, Haloplanus, Natronomonas, Desulfobacter, Haloterrigena, Desulfovibrio, Desulfococcus, Staphylococcus, Pseudomomnas, Halolamina, Halosimplex, Desulfoarcina, Peptococcus, Marinobacter, Rubrobacter and Salinibacter were identified (Fig.
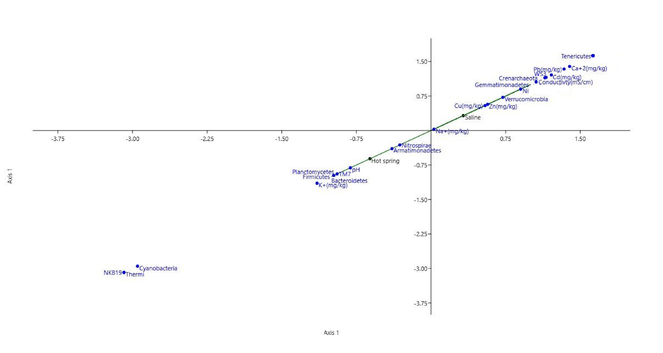
Canonical Correspondence Analysis was performed to study the effect of physicochemical parameters on bacterial communities. In the saline sample, species richness and abundance was attributed to the electrical conductivity, Na+, Ca+2 and other heavy metal concentrations. In the hot springs sediment sample, pH and K+ concentration are crucial in shaping bacterial community (Fig.
Discussion
Microbial community structure and function can be efficiently demonstrated by their microbial diversity (
Bacterial diversity of hot spring sediments detected an abundance of Firmicutes and Proteobacteria. Our results are consistent with
Prokaryotic halophiles are ubiquitous in nature and have been studied using both culture-based techniques and culture-independent sequence-based approaches. Sequence-based environmental metagenomic studies are rapidly increasing the existing knowledge of non-cultivable microbial communities, such as halophilic bacteria and archaea (
Euryarchaeota was second most abundant phylum in saline sample. The archaeal domain is a third line of evolutionary lineage varying from bacteria and eukarya. Most of studies limit archaea to extreme environmental conditions (
Predominant genera, identified in the present study, include Haloarcula, Halorhabdus, Halorubrum, Alkalibacillus, Halomicronema, Halomonas, Rhodobaca, Salsuginibacillus and Haloterrigena. Members of the genus Haloarcula are Gram-negative, rods or pleomorphic archaea commonly found in hypersaline environments, such as saline soils, salt samples and salt lakes, salterns. They are usually pigmented, neutrophils and extremely halophilic i.e. grow at 1.7–5.2 M sodium chloride (NaCl). Nine species of the genus, identified so far, include H. vallismortis, H. marismortui, H. amylolytica, H. tradensis, H. hispanica, H. japonica, H. salaria, H. quadrata and H. argentinensis (
Conclusion
The present study concluded that extreme sites of Pakistan are rich in prokaryotic diversity. Major phyla identified in hot spring samples were poly-extremophiles and have been adapted to more than one extreme conditions. Diversity analysis of saline metagenomes showed abundance of Proteobacteria as a major phylum of halophilic community. Moreover, environmental factors are playing key roles in shaping extremophilic microbial communities. Overall, this study reflected the both culturable and non-culturable prokaryotic diversity of unexplored extreme habitats of Pakistan which can be exploited further for the discovery of novel biomolecules having industrial significance.
Data Archiving Statement
The original sequencing output files have been deposited in the Sequence Read Archive (SRA) service of the National Centre for Biotechnology Information (NCBI) database under the accession numbers SAMN08026743 and SAMN08026744, respectively.
Acknowledgements
This research did not receive any specific funding.
Hosting institution
Department of Biotechnology. Fatima Jinnah Women University, Rawalpindi, Pakistan
Author contributions
AF and AY conceived and designed the experiments, AF conducted experiments, drafted the original manuscript and analysed the results, AY administered the project and edited the manuscript. SJ edited and reviewed manuscript. JCB edited the manuscript and analysed the results. All authors reviewed the manuscript.
Conflicts of interest
None
References
- Polyphasic characterization of benthic, moderately halophilic, moderately thermophilic cyanobacteria with very thin trichomes and the proposal of Halomicronema excentricum gen. nov., sp. nov.Archives of Microbiology177:361‑370. https://doi.org/10.1007/s00203-001-0390-2
- Diversity and distribution of thermophilic bacteria in hot springs of Pakistan.Microbial Ecology74.
- Halorhabdus Bergey's manual of systematics of Archaea and Bacteria. In:Bergey's Manual of Systematics of Archaea and Bacteria. https://doi.org/10.1002/9781118960608.gbm01349
- Salinity and bacterial diversity: to what extent does the concentration of salt affect the bacterial community in a saline soil?PLOS One9.
- QIIME allows analysis of high-throughput community sequencing data.Nature Methods7:335‑336. https://doi.org/10.1038/nmeth.f.303
- Archaeal habitats from the extreme to the ordinary.Canadian Journal of Microbiology52.
- Diversity of thermophiles in a Malaysian hot spring determined using 16S rRNA and shotgun metagenome sequencing.Frontiers in Microbiology6.
- Comparative analysis of microbial diversity in two hot springs of Bakreshwar, West Bengal, India.Genomics Data12.
- Microbiome Helper: a custom and streamlined workflow for microbiome research.mSystems2.
- Evaluation of wet digestion methods for quantification of metal content in electronic scrap material.Resources6.
- Halophiles. In:eLS.John Wiley & Sons, Ltd
- Genotypic and lipid analyses of strains from the archaeal genus Halorubrum reveal insights into their taxonomy, divergence, and population structure.Frontiers in Microbiology9.
- Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP).BMC Microbiology8.
- Space microbiology.Microbiology and Molecular Biology Reviews74.
- Thermophilic bacteria from the hot springs of Gilgit (Pakistan).Journal of Animal and Plant Sciences22.
- Microbial diversity in water and sediment of Lake Chaka, an athalassohaline lake in northwestern China.Applied and Environmental Microbiology72.
- Halophilic Prokaryotes in Urmia Salt Lake, a Hypersaline Environment in Iran.Curr Microbiolhttps://doi.org/10.1007/s00284-021-02583-w
- Bacteria and Archaea diversity within the hot springs of Lake Magadi and Little Magadi in Kenya.BMC Microbiology16.
- Comparison of next-generation sequencing systems.Journal of Biomedicine and Biotechnologyhttps://doi.org/10.1007/978-1-4939-7471-9_12.
- Extremophilic bacteria and microbial diversity.Annals of the Missouri Botanical Garden87.
- Proteobacteria. In:Encyclopedia of Astrobiology.Springerhttps://doi.org/10.1007/978-3-642-11274-4_1288
- Metagenomics and Culture-Based Diversity Analysis of the Bacterial Community in the Zharkent Geothermal Spring in Kazakhstan.Curr Microbiolhttps://doi.org/10.1007/s00284-021-02545-2
- Meta-analysis of microbial communities in hot springs: Recurrent taxa and complex shaping factors beyond PH and temperature.Microorganisms8(6). https://doi.org/10.3390/microorganisms8060906
- Extremophiles and extreme environments.Life3.
- Spatial algorithms applied to landscape diversity estimate from remote sensing data. In:Developments in Environmental Modelling.25.Elsevierhttps://doi.org/10.1016/B978-0-444-59396-2.00023-7
- Estimating coverage in metagenomic data sets and why it matters.The ISME Journal8.
- Hot springs of Indian Himalayas: Potential sources of microbial diversity and thermostable hydrolytic enzymes.3 Biotech7.
- Abundance, composition, diversity and novelty of soil Proteobacteria.The ISME Journal3.
- Cultivation-based and molecular assessment of bacterial diversity in the rhizosheath of wheat under different crop rotations.PLOS One10https://doi.org/10.1371/journal.pone.0130030
- Alkalibacillus halophilus sp. nov., a new halophilic species isolated from hypersaline soil in Xin-Jiang province, China.Systematic and Applied Microbiology30.
- Environmental factors driving spatial heterogeneity in desert halophile microbial communities.Frontiers in Microbiology11.
- Haloarcula. In:Bergey's Manual of Systematics of Archaea and Bacteria. https://doi.org/10.1002/9781118960608.gbm00481
- Salinity shapes microbial diversity and community structure in surface sediments of the Qinghai-Tibetan Lakes.Scientific Reports6.
- Phylogenetic diversities and community structure of members of the extremely halophilic Archaea (order Halobacteriales) in multiple saline sediment habitats.Applied and Environmental Microbiology78.
- Comparative molecular analysis of chemolithoautotrophic bacterial diversity and community structure from coastal saline soils, Gujarat, India.BMC Microbiology12.